 JA
JA
 EN
EN
 中文
中文



Miriam Coelho Molck Milena Simioni Társis Paiva Vieira
Fabíola Paoli Monteiro Vera L. Gil-da-Silva-Lopes
Department of Genetics, Faculty of Medicine, University of Campinas (UNICAMP), Campinas, Brazil
Confirmed facts
The reported 10q22.3q23.2 microdeletion is associated with the recently described rare 10q22q23 deletion syndrome , which exhibits a non-characteristic phenotype.
To date, 14 cases of microdeletions in the low-copy repeat 3/4 flanking 10q22.3q23.2 have been reported in the literature.
New Insights
We report a novel case of a boy with a 7.8 Mb microdeletion at 10q22.3q23.2 and compare his clinical manifestations with similar cases reported in the literature.
In addition, our patient has a 189 kb 16q12.1 microdeletion that may be of phenotypic relevance.
keyword
Deletion syndrome 10q22q23 – Genotype-phenotype correlations – Molecular cytogenetics
summary
Deletions of the 10q22.3q23.2 region are rare and are mediated by two low-copy repeats (LCRs 3 and 4). These deletions have already been recognized as the 10q22q23 deletion syndrome . The phenotype associated with this condition is rather uncharacteristic, with the most common features being craniofacial dysmorphism and developmental delay. We report a boy with craniofacial dysmorphic features, developmental delay, tetralogy of Fallot, limb anomalies, and recurrent respiratory tract infections. Chromosomal microarray analysis showed a 7.8 Mb microdeletion in 10q22.3q23.2 , adjacent to LCRs 3/4, and a further 189 kb microdeletion in 16q12.1 . In this paper, we review the clinical manifestations of reported cases with similar deletions and compare them with our patient, contributing to a better understanding of genotype-phenotype correlations.
Copyright (2017 S. Karger AG, Basel)
Improved molecular cytogenetic analysis. With the advancement of molecular cytogenetic analysis using MLPA and chromosomal microarrays, the list of microdeletion / microduplication syndromes is constantly growing [Slavotinek, 2008; Nevado et al. 2014]. Some of these have recognizable phenotypes, while others are more phenotypically inconsistent and confer increased risk for neuropsychiatric disorders [Koolen et al. 2008; van Bon et al. 2011].
Deletions of 10q22.3q23.2 have been characterized as a rare novel syndrome associated with cognitive and behavioral disorders [Balciuniene et al., 2007]. This region is flanked by a complex set of low copy repeats (LCRs), designed LCR3 and LCR4, that can cause a variety of genomic alterations mediated by nonallelic homologous recombination (NAHR) [Lupski and Stankiewicz, 2005].
The presence of LCRs in this region suggests that this locus is susceptible to chromosomal rearrangements [Alliman et al. 2010]. Nevertheless, only 14 de novo deletions and seven duplications with breakpoints within LCR3/4 have been described [Goss et al. 1998; Han et al. 2004; Dufke et al. 2006; Balciuniene et al. 2007; Erdogan et al. 2008; Alliman et al. 2010; Reddy et al. 2011; Singh et al. 2011; van Bon et al. 2011; Petrova et al. 2014].
Here we report a patient with a 7.8 Mb microdeletion at 10q22.3q23.2 adjacent to LCR3/4 and an additional 189 kb microdeletion at 16q12.1 .
Patients and methods
Case report
A 3-year-old boy was referred for medical genetic consultation at the Genetics Service of FCM-UNICAMP because of multiple anomalies. The patient was evaluated by an experienced clinical geneticist using a specific checklist of clinical and family history information and dysmorphic features.
The proband was the second child born to unrelated parents with an older, healthy child, who had previously had a spontaneous abortion. His family history was unremarkable. At birth, he had bilateral clubfeet. Tetralogy of Fallot was diagnosed in the neonatal period, and he underwent corrective cardiac surgery at 9 months of age. His development was generally delayed thereafter, with one reported febrile seizure during childhood, and he had recurrent upper respiratory tract infections.
Morphologic examination at the age of 3 years revealed a frontal boss, low-set anteverted ears with bilateral overfolded helices and prominent right antihelix, right preauricular fossa, sparse eyebrows, inferiorly oblique palpebral fissures, shallow orbits, bulbous tip of the nose, high-arched palate, bilateral club feet, and bilateral incomplete palmar clefts.
Molecular cytogenetic analysis
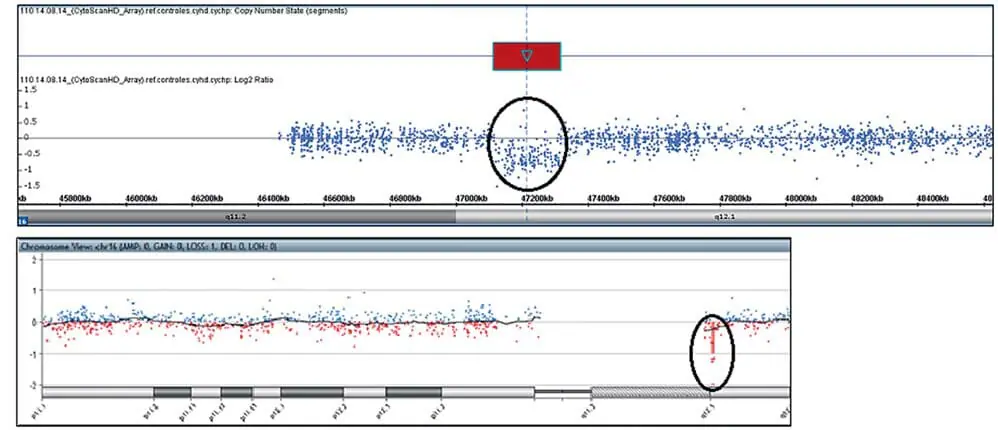
Chromosomal microarray analysis was performed using a CytoScan HD chip (Affymetrix, Santa Clara, CA, USA) according to the manufacturer’s instructions and analyzed using the Chromosome Analysis Suite (ChAS) software (Affymetrix). Confirmation of the pathogenic CNV was performed using an Agilent Human Genome G3 SurePrint 8 × 60 K Microarray (Agilent Technologies, Santa Clara, CA, USA) and analyzed using the Agilent Cytogenomics software (Agilent Technologies). Chromosomal microarray analysis of the patient revealed a 7.8 Mb microdeletion at 10q22.3q23.2 ( Figure 1A,B ). In addition, our patient had a 189 kb microdeletion in 16q12.1 (Figure 2A,B) : arr [GRCh37] 10q22.3q23.2(8146577_89253430) × 1, 16q12.1(47127281_47316339) × 1. Investigation of the parental origin of these deletions was not possible due to unavailability of samples. The patient was lost to follow-up and a karyotype could not be performed to exclude structural alterations.
Figure 1. Array results for affected individuals. Chromosomal microarray analysis (CMA) hybridization profile of chromosome 10 using the CytoScan HD chip showed a 7.8 Mb microdeletion at q22.3q23.2 (81,446,577-89,253,430; hg 19) . CMB hybridization profile of chromosome 10 using the Agilent Human Genome G3 SurePrint 8×60 K microarray confirmed the microdeletion at q22.3q23.2.
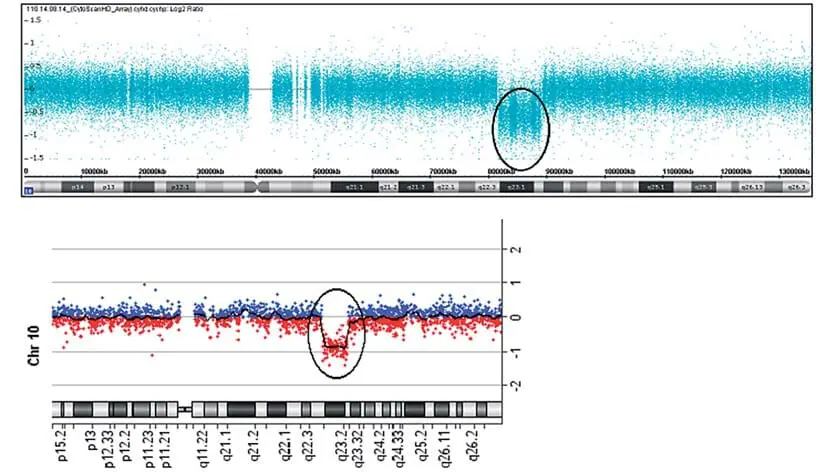
Figure 2. Sequencing results for affected individuals. Chromosomal microarray analysis (CMA) hybridization profile of chromosome 16 using CytoScan HD chips showed a 189 kb microdeletion at q12.1 (47,127,281-47,316,339; hg 19) . CMB hybridization profile of chromosome 16 using Agilent Human Genome G3 SurePrint 8×60 K microarrays confirmed the q12.1 microdeletion .
Observations
We report the case of a male infant with developmental delay, congenital heart disease (CHD), craniofacial dysmorphism, limb anomalies, and recurrent infections. Indeed, the patient presents with a 7.8 Mb 10q22.3q23.2 interstitial microdeletion overlapping the rare 10q22q23 deletion syndrome (DS) region. Common clinical features of the reported patient and proband with the LCR3/4 10q22.3q23.2 deletion included craniofacial dysmorphic features present in all 15 cases, developmental delay (13/15, 87%), palate alterations (6/15, 40%), congenital heart disease (6/15, 40%), and limb anomalies (10/15, 67%). Other rare clinical features of 10q22q23 DS not present in our patients were hypotonia (3/15, 20%), autism (2/15, 13%), and joint hyperextensibility (2/15, 13%) ( Table 1 ). Craniofacial dysmorphisms did not result in a recognizable facial phenotype and most commonly included hypo- or hyperphthalmos (9/15, 60%), low-set ears and small ears (6/15, 40%), upturned or downturned palpebral fissures (6/15, 40%), upturned folds (4/15, 27%), and flat nasal bridge (3/15, 20%). Our patients presented with low-set ears and downturned palpebral fissures. The most common palatal alteration was a high-set palate, seen in 5 of 15 cases (33%), including the one reported in this paper. The most frequently observed congenital heart defect was ventricular septal defect, identified in 3 of 15 patients (20%), although tetralogy of Fallot was not present in our patient. Hand and foot anomalies commonly included clubfoot (4/15, 27%), which was also present in our patient. Arachnodactyly was also included (2/15, 13%). Another clinical feature observed in our patient was recurrent upper respiratory tract infections, which were not reported in other 10q22q23 DS cases. There are 16 OMIM genes within the 7.8 Mb deletion region at 10q22.3q23.2 detected in our patient. Some of these genes, e.g., BMPR1A (601299), LDB3 (605906), GRID1 (610659), and NRG3 (605533), have been hypothesized as putative candidate genes related to the phenotype, especially with regard to neuropsychological development and cardiac defects [Balciuniene et al. 2007; van Bon et al. 2011; Breckpot et al. 2012]. Saito et al. [2012] evaluated the cardiac morphology of mutant mice knockout BMPR1A protein in neural crest-derived cells and showed that some of them showed facial fusion defects such as cleft face and cleft palate, or facial dysmorphic features besides cardiac defects. Thus, the craniofacial dysmorphism and congenital heart defects observed in our patient may have been partially caused by the deletion of the BMPR1A gene.
Heterozygous point mutations or partial deletions of the BMPR1A gene are also associated with juvenile polyposis syndrome (OMIM 174900), characterized by the development of hamartomatous polyps in the gastrointestinal tract [Larsen and Howe, 2017]. Among patients with 10q22.3q23.2 deletions , one subtype of juvenile polyposis syndrome, called infantile juvenile polyposis syndrome, has been observed only in patients with deletions of both the BMPR1A and PTEN genes [Menko et al., 2008]. PTEN has a recognized tumor suppressor role [Li et al., 1997] and has been associated with Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome, suggesting that sequential deletions of both BMPR1A and PTEN are required for polyposis development [Zhou et al., 2003; Dahdaleh et al., 2012]. PTEN is absent in LCR 3/4 flanking 10q22.3q23.2 deletions , and no patients with similar deletions have been reported to develop juvenile polyposis or a polyposis phenotype in infancy [Petrova et al 2014].
LDB3 encodes the LIM-binding domain 3 protein, which is involved in cytoskeletal assembly, and mutations in this gene are associated with dilated cardiomyopathy in humans [Vatta et al. 2002; Hershberger et al. 2008]. The effects of this protein on cardiac function or structure are under investigation [Aliman et al. 2010].
The GRID1 gene encodes a subunit of glutamate receptor channels and plays a role in mediating excitatory synaptic transmission in the central nervous system [Yamazaki et al., 1992]. Vasan et al. [2009] proposed GRID1 as a candidate gene for left ventricular wall thickness through a genome-wide association meta-analysis of common genetic variants associated with cardiac structure and function. Furthermore, this gene has also been associated with schizophrenia in several studies [Fallin et al., 2005; Coyle, 2006; van Bon et al., 2011]. Another gene located proximal to GRID1 and reported to be associated with schizophrenia risk and symptoms is NRG3 [Chen et al., 2009]. NRG3 plays a role in signaling proteins that mediate cell-cell interactions in the nervous system, heart, breast, and other organ systems [Falls, 2003].
In addition, our patient has a microdeletion in 16q12.1 (189 kb) . This region contains two OMIM genes: NETO2 (607974) and ITFG1 (611803). NETO2 is a kainate receptor subunit that is thought to have a significant effect on glutamate signaling in the brain [Zhang et al. 2009]. ITFG1 is a transmembrane protein with proposed cell adhesion functions [Kato et al. 2014]. This deletion was not detected in any individual in the DGV database (http://projects.tcag.ca/variation). In the DECIPHERDatabase (https://decipher.sanger.ac.uk/), a much larger deletion that encompasses the deletion found in our case ) is described as the only finding found in two cases (2856 and 289229), with developmental delay in common with our case. Also, case 289229 from DECIPHER presented with a congenital heart defect, an atrial septal defect. To clarify the significance of this deletion on the phenotype, more detailed information on other individuals with similar deletions is needed, especially on the specific functions of these genes. In addition, 3 of 15 10q22q23 DS patients had chromosomal abnormalities ( Table 1 ), which may have influenced the phenotype. Recently, it has been suggested that large genome-scale structural variants, i.e., changes involving large bases of DNA, may be associated with further complexity, such as other structural variants generated in one-off events (chromoanasynthesis and chromothripsis-like events) or complex genomic rearrangements with loss of heterozygosity [Carvalo and Lupski, 2016]. Although different disease phenotypes affect different organ systems, overlapping disease phenotypes are likely caused by two genes encoding interacting proteins within the same pathway. This interplay between pathogenic variants in multiple genes results in the complex spectrum of phenotypes observed within an individual and contributes to the variability of associated genomic disorders [Posey et al. 2017].
Most of the clinical manifestations noted in our patient have been reported in the rare 10q22q23 DS. However, the findings in our patient and others with this deletion do not allow characterization of the core phenotype. Furthermore, an influence of the 16q12.1 deletion on the phenotype cannot be excluded.
The identification of rare and novel genomic imbalances in young patients could change clinical management by allowing early intervention for behavioral abnormalities and developmental delays and ensure specific follow-up. Additional cases would improve genotype-phenotype correlations and the effect of multiple rare variants on the pathogenesis of the disease.
Table 1 Clinical characteristics of patients with 10q22.3q23.3 deletion adjacent to LCR3/4
| References, cases | Deletion size | Inheritance | Gender | Age at time of evaluation | Developmental delay | Congenital heart disease | Palate changes | Maxillofacial dysmorphia | Limb changes | < th>Other clinical featuresOther malformations | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Barciunien et al. [2007] | |||||||||||
| UM10qDel-01 | 7.5 Mb | de novo | Male | 3 years and 6 months old | + | – | – | < td>Mild features– | Autism | – | |
| JHU10qDel-01 | 7.5 Mb | de novo | Male | 1 year and 6 months | + | – | – | < td>white anterior lock– | posterior cerebellum, microcyst, cerebellum | – | |
| Aliman et al. [2010] | |||||||||||
| Case 1 | 7.25 Mb | de novo | Male | 2 years 7 months | + | Patent ductus arteriosus | High arcuate Palate | Mild hyperpigmentation, erect Raised palpebral fissures, excessive folding of the lateral pinna, wrinkled ear lobes, thin upper lip, micrognathia Hyperactivity | – | ||
| Case 2 | 7.25 Mb | de novo | Female | 17 years 1 month | + | – | High arched palate | Mild facial asymmetry, downward palpebral fissures, border Borderline set ears, flattened cheeks, prominence | Mild arachnodactyly | Mild hypotonia, joint hyperextensibility, history of myopia, attention deficit/ Hyperactivity disorder | – |
| Case 3 | 7.25 Mb | de novo | Male | 8 days | NA | – | – | Mild Plagiocephaly, blepharophaly, mild Normocephaly with severe ocular folds, low-set, backward-rotated ears, and narrow external auditory canals | Bilateral clubfeet | Mild hypotonia, bilateral hearing loss | – |
| Case 4 | 7.25 Mb | de novo | Male | 1 year 8 months | + | – | High arched palate | Macrocephaly, frontal bossing , narrow vertex, retrognathia, microstomia, lower oblique palpebral fissures, hyperpigmentation, occipital occipitalis, bilateral notch intermediate spiral | – | mild hypotonia, Failure to thrive | – |
| van Bonら [2011] | |||||||||||
| Patient 1 | 7.2 Mb | de novo | Female | Over 22 years old | + | – | – | Decreased peripheral function, orthostatic palpebral fissures, ptosis, low-set microtia | platysma and hallux | breast hypoplasia | – |
| Patient 2 | 7.2 Mb | de novo | Female | 2 years 6 months | + | Atrial ventricular septal defect | –< /td> | High eyelids, anteverted nostrils, flat nasal bridge, large mouth, telecanthosis, low-set ears | – | – | – |
| Patient 3 | 7.7 Mb | de novo | Male | 3 years 7 months old | + | – | High arched palate | Dolichocephali -, high eyelids, upper eyelid folds, flat nasal bridge, sunken midface, low-set ears | posterior thumb | malignant teeth, Chiari I malformation, epilepsy< /td> | de novo 2q36.3 replication (722 kb) |
| Patient 4 | 7.7 Mb | de novo | Male | 12 years old | + | TR, pulmonary valve regurgitation | – | Long face, Hyperorism, almond-shaped eyes | Radioulnar fusion, scoliosis, kyphosis, pectus excavatum | Café au lait spots | 47,XYY |
| Patient 5 | 7.5 Mb | Maternal origin | Male | 5 years old | + | – | – | High eyelids, wide base of the nose, flat nasal bridge, low-set ears | Bilateral clubfeet | Short scrotum | – |
| Reddy et al. [2011] | |||||||||||
| Case 2 | 7.7 Mb | de novo | Female | 4 years old | + | – | – | wide nose, inverted lower lip, green border | – | – | – |
| Singh et al. [2011] | |||||||||||
| Case report | 7.46 Mb | de novo | Female | 5 years 6 months | + | Atrial ventricular septal defect | –< /td> | Mild canopy cleft, rightward exotropia | Hyperkeratosis, fifth toe nail | Persistent, tachypnea, recurrent otitis media Otitis media | – |
| Petrova et al. [2014] | |||||||||||
| Clinical report | 7.3 Mb | de novo | Male | 2 years old | – | Atrial ventricular septal defect, persistent foramen ovale | Median cleft palate | High eyelids, high eyelids, wide nasal bridge, protruding palpebral fissures, maxillary folds, posterior jaw, ptosis | Right side clubfoot | – | – |
| Current case | 7.8 Mb | Unknown | Male | 3 years old | + | Tetralogy of Fallot | < td>High arcuate palateFrontal process, low and slightly anteverted pinnae, excessive bilateral Folded helix, prominent right antihelix, right preauricular fossa, mild eyebrows, lower oblique palpebral fissures, superficial orbits, tip of the nasal bulb | Bilateral clubfeet, bilateral incomplete palmar Wrinkles | Recurrent upper respiratory tract infections | 16q12.1 deletion (189 kb) | |
Acknowledgements
We thank the patients and their families for their collaboration and the multi-user instrumentation laboratory at the Laboratory of Molecular Genetics, School of Medicine, University of Campinas, Campinas, São Paulo, Brazil, for their assistance with the equipment, GeneChip Fluidics Station, and GeneChip Scanner.
This work was supported by FAPESP – Fundaco de Amparo to Pesquissa do Estado Sano Paulo (2008/10596-0, 2009/08756-1, 2011/23794-7, 2012/51799-6) and CNPq – Conselho Desolvimento Cientifico Technologico (149600/2010-0, 471422/2011-8). VLG-d.-S.-L. is supported by CNPq (30455/2012-1).
Ethics Statement
The study was approved by the Ethics Committee of the University of Campinas (No. 487/2009 and 433/2010). Written informed consent was obtained from the patient’s parents. Disclosure statement The authors declared that they had no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
Alliman S, Coppinger J, Marcadier J, Thiese H, Brock P, et al: Clinical and molecular characterization of patients with recurrent genomic disorders at 10q22.3q23.2. Clin Genet 78: 162-168 (2010)
Balciunien J, Feng N, Iyadurai K, Hirsch B, Sharnas L et al.: Recurrent 10q22-q23 deletion: a genomic disorder on 10q associated with cognitive and behavioral abnormalities. Am J Hum Genet 80: 938–947 (2007).
Breckpot J, Tranchevent LC, Thienpont B, Bauters M, Troost E, et al: BMPR1A is a candidate gene for congenital heart disease associated with recurrent 10q22q23 deletion syndrome. Eur J Med Genet 55: 12–16 (2012).
Carvalho CM, Lupski JR: Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17: 224–238 (2016). Chen PL, Avramopoulos D, Lasseter VK, Mc-Grath JA, Fallin MD, et al: Fine mapping on chromosome 10q22-q23 implicates neuregulin 3 in schizophrenia. Am J Hum Genet 84: 21–34 (2009).
Coyle JT: Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol 26: 365–384 (2006).
Dahdaleh FS, Carr JC, Calva D, Howe JR: Juvenile polyposis and other intestinal polyposis syndromes associated with microdeletions of chromosome 10q22-23. Clin Genet 81: 110-116 (2012)
Dufke A, Singer S, Borell-Kost S, Stotter M, Pflum DA, et al: De novo structural chromosomal imbalances: molecular cytogenetic characterization of partial trisomies. Cytogenetics Genome Research 114:342-350(2006).
Erdogan F, Belloso JM, Gabau E, Ajbro KD, Guitart M, et al: Fine mapping of a novel interstitial 10q22-q23 duplication in a patient with congenital heart disease and microcephaly.
Eur J Med Genet 51: 81–86 (2008).
Fallin MD, Lasseter VK, Avramopoulos D, Nicodemus KK, Wolyniec PS, et al: Bipolar I disorder and schizophrenia: a 440-single nucleotide polymorphism screen of 64 candidate genes in an Ashkenazi Jewish caseparent trio. Am J Hum Genet 77: 918–936 (2005).
Falls DL: Neuregulins: function, morphology, and signaling strategies. Cell Exp 284: 14-30 (2003)
Goss PW, Voullaire L, Gardner RJ: Intrachromosomal insertion duplication 10q22.1eq25.1: second case. Ann Genet 41: 161-163 (1998)
Han JY, Kim KH, Jun HJ, Je GH, Glotzbach CD, Shaffer LG: Partial trisomy(15;10) of chromosome 10 (q22-q24) due to a maternal insertion translocation. Am J Med Genet A 131: 190–193 (2004).
Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, et al.: Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci 1: 21–26 (2008).
Kato M, Chou TF, Yu CZ, DeModena J, Sternberg PW: LINKIN is a novel transmembrane protein required for cell adhesion. 3:e04449 (2014).
Koolen DA, Sharp AJ, Hurst JA, Firth HV, Knight SJ, et al: Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J Med Genet 45: 710-720 (2008).
Larsen HJ, Howe JR: Juvenile polyposis syndromes, in Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, et al (eds): GeneReviews® [Internet] (University of Washington, Seattle 1993-2017). First published: May 13, 2003, last updated: December 3, 2015.
Li J, Yen C, Liaw D, Podsypanina K, Bose S, et al: PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancers. Science 275:1943-1947(1997).
Lupski JR, Stankiewicz P: Genomic disorders: molecular mechanisms of rearrangements and transmitted phenotypes. PLoS Genet 1:e49 (2005).
Menko FH, Kneepkens CM, de Leeuw N, Peeters EA, Van Maldergem L, et al: Variable phenotypes associated with 10q23 microdeletions involving the PTEN and BMPR1A genes. Clin Genet 74: 145-154 (2008).
Nevado J, Mergener R, Palomares-Bralo M, Souza KR, Vallespin E, et al: New microdeletion and microduplication syndromes: a comprehensive review. Genet Mol Biol 37 Suppl 1: 210– 219 (2014).
Petrova E, Neuner C, Haaf T, Schmid M, Wirbelauer J, et al: A boy with an LCR3/4-contiguous 10q22.3q23.2 microdeletion and rare phenotypic features. Mole Syndrome 5: 19-24 (2014)
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, et al: Uncovering disease phenotypes caused by multilocus genomic variants. N Engl J Med 376: 21-31 (2017)
Reddy KS, Mardach R, Bass H: Oligoarray (105K) CGH analysis of a chromosomal microdeletion within 10q22.1q24.32. Cytogenetics Research 132:113-120(2011).
Saito H, Yamamura KI, Suzuki N: Reduced bone morphogenetic protein receptor type 1A signaling in neural crest-derived cells leads to facial dysmorphism. Disc Model Mech 5: 948-955 (2012)
Singh S, Aftimos S, George A, Love DR: Interstitial deletions of 10q23.1 and identification of three 10qdel syndromes. Singapore Med J52:e143-146(2011)
Slavochnek AM: A novel microdeletion syndrome detected by chromosomal microarray. Hum Genet 124: 1-17 (2008).
van Bon BW, Balciuniene J, Fruhman G, Nagamani SC, Broome DL, et al: Phenotype of recurrent 10q22q23 deletions and duplications.
Eur J Hum Genet 19: 400–408 (2011).
Vasan RS, Glazer NL, Felix JF, Lieb W, Wild PS, et al: Genetic variants associated with cardiac structure and function: meta-analysis and replication of genome-wide association data. JAMA 302:168-178(2009)
Vatta M, Stetson SJ, Perez-Verdia A, Entman ML, Noon GP, et al: Molecular remodeling of dystrophin in patients with end-stage cardiomyopathy and recovery in patients receiving support device therapy. Lancet 359: 936-941 (2002)
Yamazaki M, Araki K, Shibata A, Mishina M, et al.: Molecular cloning of a cDNA encoding a novel member of the mouse glutamate receptor channel family. Biochem Biophys Res Commun 183: 886–892 (1992).
Zhang W, St-Gelais F, Grabner CP, Trinidad JC, Sumioka A, et al: Transmembrane minor subunits modulate kainate glutamate receptors. Neuron 61:385-396(2009).
Zhou XP, Waite KA, Pilarski R, Hampel H, Fernandez MJ, et al: Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in abnormal PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am J Hum Genet 73: 404–411 (2003).







