
 JA
JA
 EN
EN
 中文
中文





ユアン-ハレル-ルプスキ症候群は、17番染色体の一部が重複することによって発生する稀な神経疾患です。ポトツキ-ルプスキ症候群やシャルコー-マリー-トゥース病の特徴を併せ持ち、発達遅延、低筋緊張、顔の特徴、末梢神経障害など多岐にわたる症状が見られます。診断と管理について解説します。

ダウン症の検査
気になる費用はこちら
この記事のまとめ
ユアン-ハレル-ルプスキ症候群(Yuan-Harel-Lupski Syndrome)は、17番染色体の一部が重複することによって引き起こされる稀な疾患で、ポトツキ-ルプスキ症候群やシャルコー-マリー-トゥース病に類似した特徴を示します。低筋緊張、発達遅延、末梢神経障害、心臓や腎臓の異常など多様な症状が見られるため、早期診断と包括的なケアが重要です。
病気の別称
- PMP22-RAI1連続遺伝子重複症候群(PMP22-RAI1 contiguous gene duplication syndrome)
- 17p11.2p12 微小重複症候群(17p11.2p12 microduplication syndrome)
- Dup(17)(p11.2p12)
- Trisomy 17p11.2-p12
- Trisomy 17p11.2p12
- Yuan-Harel-Lupski syndrome
疾患概要

ユアン-ハレル-ルプスキ症候群(Yuan-Harel-Lupski Syndrome, YUHAL)は、17番染色体の長腕の一部が部分的に重複することによって引き起こされる稀な神経疾患です。この疾患は、ポトツキ-ルプスキ症候群(Potocki-Lupski Syndrome)と1A型シャルコー-マリー-トゥース病(Charcot-Marie-Tooth disease, CMT1A)の特徴を組み合わせた症状を示します。
YUHAL症候群の最初の兆候は通常、乳児期に現れます。影響を受けた乳児はしばしば筋肉の緊張が弱い状態(低筋緊張)を示し、これにより摂食困難や成長障害(体重や身長の増加不良)を引き起こすことがあります。運動スキル(歩行など)や言語スキルの発達が遅れることが一般的で、行動面の問題、例えば睡眠時無呼吸や入眠および睡眠の維持が難しいなどの睡眠問題が頻繁に見られます。また、下向きの目の角度(下斜眼裂)、三角形の顔立ち、目が揃わない(斜視)など、微妙な顔立ちの特徴が見られることがあります。これらの特徴はポトツキ-ルプスキ症候群に見られるものと似ています。
幼少期には、末梢神経の損傷に関連する追加の症状が現れます。末梢神経は、脳や脊髄から筋肉や感覚細胞(触覚、熱、冷たさなどを感じる細胞)をつなぐ役割を果たしています。この神経が徐々に損傷されることで、特に下肢の筋力低下が進行し、歩行困難や異常な歩行スタイルを引き起こす場合があります。扁平足(ペスプラヌス)、高アーチ足(ペスカバス)、内反足(内側や上方に向いた足)などの足の異常も一般的です。他の症状として、反射の減少または消失、足や下肢における触覚や温度感覚の低下、筋肉の萎縮(アトロフィー)が含まれます。これらの症状はCMT1Aに見られるものと似ていますが、YUHAL症候群ではしばしば5歳未満で発症することが特徴です。
YUHAL症候群では、心臓や腎臓などの組織や臓器の異常も見られることがあります。顔面の形態異常(顔面異形成)が観察されることもあり、稀なケースでは、脊髄内に液体がたまる嚢が形成される疾患である脊髄空洞症(シリンゴミエリア)が発生することもあります。
YUHAL症候群は、複数の身体システムに影響を及ぼす多様な症状を特徴としており、早期診断と多職種による包括的なケアが必要です。
病因と診断の方法
連続遺伝子症候群とは、染色体の異常によって複数の隣接した遺伝子が影響を受けることで引き起こされる遺伝性疾患です。これらの異常は、染色体上の遺伝子の配置やコピー数(量)を乱し、遺伝子が正しく機能しなくなったり、遺伝子のコピー数が多すぎたり少なすぎたりする原因となります。このような症候群は、通常稀で、遺伝することはほとんどなく、症状や重症度に幅広い変化が見られる(高い多様性がある)ことが特徴です。1つの遺伝子に小さな変化(点突然変異)が生じることで発症する遺伝病とは異なり、連続遺伝子症候群はDNAの大きなセクションが再編成されることで発症します。このため、これらは「ゲノム障害」として分類されます。ゲノム障害とは、遺伝子全体の規模で変化が起こる疾患を指します。
こうした再編成の一般的な結果の1つとして、コピー数変異(CNV)の形成があります。CNVは、DNAの構造的な変化のことで、DNAの一部が複製(余分なコピーが作られる)されたり、削除(コピーが欠損する)されたりします。これらの変異は顕微鏡で見るには小さすぎますが、遺伝子の正常な機能を妨げるほどの大きさがあります。CNVのサイズは数千塩基対(キロベース、kb)から数百万塩基対(メガベース、Mb)までさまざまであり、通常の状態(各遺伝子が両親から1コピーずつ合計2コピー持つ状態)からの偏りを引き起こします。

連続遺伝子症候群の一例として、ポトツキ-ルプスキ症候群(Potocki-Lupski Syndrome, PTLS)に関連するゲノム重複と、シャルコー-マリー-トゥース病1A型(Charcot-Marie-Tooth disease type 1A, CMT1A)に関連するゲノム重複があります。PTLSは、低筋緊張(筋肉の緊張が弱い状態)、成長不良(failure to thrive)、体重の減少、知的障害、自閉症に関連する特徴を示します。一方、CMT1Aは、末梢神経に影響を与える一般的な遺伝性疾患で、手足の筋力低下や筋肉の萎縮を引き起こします。この疾患は「常染色体優性遺伝」とされ、片方の親から変異遺伝子を1つ受け継ぐだけで発症することを意味します。
これらの疾患に関連する重要な遺伝子は、PTLSに関与するRAI1遺伝子と、CMT1Aに関与するPMP22遺伝子です。これらはどちらもコピー数に敏感な遺伝子(コピー数が正常でないと機能に問題が生じる遺伝子)です。PTLSおよびCMT1Aのほとんどの症例は、非アレリ性相同組換えと呼ばれる過程によって引き起こされる反復的な重複に起因します。この過程は、ゲノム内の特定の低コピー繰り返し領域(LCR)間で、細胞分裂中に誤った並びが起こることで発生します。しかし、RAI1とPMP22の近くにあるLCRは非常に似ているわけではないため、両方の遺伝子を含む重複は稀であり、異なる突然変異の仕組みによって引き起こされる可能性が高いと考えられています。
PTLSの発症率は約50,000人に1人と推定されており、シャルコー-マリー-トゥース病(CMT)はその遺伝的サブタイプ全体(最も一般的なCMT1Aを含む)を含めて、約2,500人に1人の割合で発症します。PTLSとCMT1Aがそれぞれ独立した出来事として発生する場合、両方を同時に持つ可能性は非常に稀で、約1億2,500万分の1と推定されています。しかし、PTLSとCMT1Aに関連する遺伝子は同じ染色体上で比較的近い場所(約2.5Mbの距離)に位置しているため、単一の突然変異によってこれら両方の遺伝子を含むDNAセグメントが重複する可能性があります。このため、両方の疾患の特徴を併せ持つ人が見られることがあります。
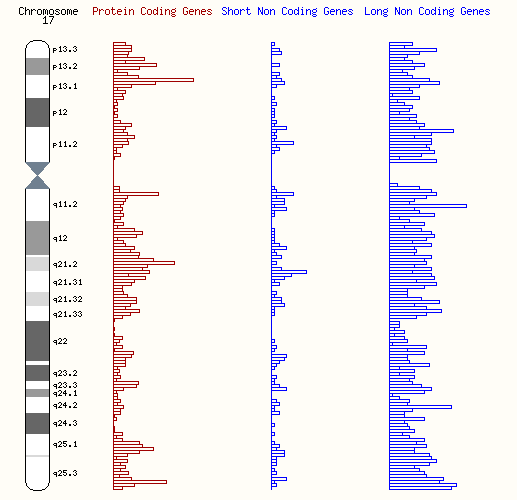
遺伝的異常を検出するための診断方法として、p11.2p12染色体領域に関連するものには、蛍光 in situ ハイブリダイゼーション(FISH)があります。これは、特定のDNA配列を識別するために蛍光プローブを使用する手法です。また、17p領域の高解像度ゲノム解析では、オリゴヌクレオチドアレイ比較ゲノムハイブリダイゼーション(aCGH)と呼ばれる方法が一般的に使用されており、遺伝物質の小さな重複や欠失を非常に高い精度で検出することができます。さらに、マイクロサテライトマーカーを使用した分子研究は、DNA内の特定のパターンを識別することで、さらなる精度を提供します。
妊娠中の診断を確定するために、羊水穿刺が使用されることもあります。この手法では、子宮から少量の羊水を採取します。しかし、DNA配列解析技術の進歩により、非侵襲的出生前検査(NIPT)が信頼性の高い安全なスクリーニング方法として導入されました。NIPTは母体の血液中に含まれる胎児のDNA断片を分析する方法であり、母体と胎児の両方にリスクを与えない代替手段として、遺伝的異常を正確に早期検出することが可能です。
疾患の症状と管理方法
この疾患に関連する症状は、身体的、発達的、神経精神的な問題を広範囲にわたって含み、その複雑な特性を反映しています。発達の遅れは、この疾患の顕著な特徴であり、その程度は軽度から重度までさまざまです。この遅れには、言語習得や全般的な知的発達の遅れなど、言語や認知における課題が含まれます。低筋緊張(筋肉の緊張が低い状態)は乳児期によく見られ、摂食困難や体重増加不良(成長障害)につながることが多いです。その他の身体的症状としては、短頭症や小頭症(頭部が短いまたは小さい状態)、低身長、顔の形態異常が挙げられます。
影響を受けた個人においてよく見られる顔の特徴には、三角形の顔、ふっくらとした頬、滑らかな人中(鼻と上唇の間の溝)、薄い上唇、不規則な形の眉、まばらな髪などがあります。他の特徴として、目尻が下向きになっている(下斜眼裂)、眼間隔が広い(眼間解離)、歯の不正咬合、深い手掌線(手のひらのしわ)が挙げられます。一部の人には、長く柔軟な指や先天性股関節脱臼、内反足(足が内側または上向きに曲がる状態)などの構造的異常が見られることもあります。

神経精神的な症状も一般的で、行動上の問題、不安、注意欠陥多動性障害(ADHD)、自閉スペクトラム症が頻繁に報告されています。一部の人は、双極性障害などのより重度の神経精神的疾患を経験することがあります。言語表現や理解が大幅に困難になるコミュニケーション障害もよく見られます。また、閉塞性睡眠時無呼吸や中枢性睡眠時無呼吸などの睡眠障害も一般的で、全体的な健康状態や認知機能を悪化させる可能性があります。
心血管系の異常もこの疾患によく関連しており、拡張した大動脈根、二尖大動脈弁、中隔欠損(心室や心房を分ける壁に穴がある状態)などの構造的異常から、継続的なモニタリングや介入が必要な機能的問題までさまざまです。
この疾患の管理には、個々の症状やニーズに合わせた多職種によるアプローチが必要です。身体療法、作業療法、言語療法を含む早期介入療法は、発達の遅れに対処し、生活の質を向上させるために重要です。心血管の異常や神経精神的な症状、その他の医学的問題を管理するために、専門医による定期的なモニタリングが必要です。摂食困難へのサポート、歯の異常に対するケア、股関節脱臼や内反足といった整形外科の問題への対応も包括的ケアの一部です。場合によっては、神経精神的症状、睡眠障害、その他の関連症状を管理するために薬物療法や行動療法が必要となることもあります。
全体として、この疾患を持つ人々は、早期診断、個別に調整された介入、多職種による連携ケアにより、発達、健康、生活の質を最適化することが可能です。
将来の見通し
ユアン-ハレル-ルプスキ症候群(Yuan-Harel-Lupski Syndrome)は非常に稀な疾患であり、影響を受けた個人の平均寿命に関するデータは限られています。しかし、関連する疾患であるポトツキ-ルプスキ症候群(Potocki-Lupski Syndrome, PTLS)やシャルコー-マリー-トゥース症候群(Charcot-Marie-Tooth Syndrome, CMT)から得られる情報が参考になります。これらの疾患はユアン-ハレル-ルプスキ症候群と共通する症状を持ちます。これらの情報から、ユアン-ハレル-ルプスキ症候群の症状はより重篤で、予後があまり良くない場合もあるものの、早期の介入や一貫したサポート、適切なケアにより、生活の質が向上し、成人期に達する可能性が示唆されています。
ポトツキ-ルプスキ症候群では、約40%の乳児が先天性心疾患を持って生まれ、その中には生命を脅かす場合もあります。それでも、PTLSの臨床記録では、30代後半まで生存した例が報告されています。同様に、シャルコー-マリー-トゥース症候群の臨床記録では、55歳まで生存した男性の例が記録されています。これらの例は、適時の医療介入と包括的なケアがこれらの疾患を管理し、寿命を延ばす上で重要であることを示しています。ユアン-ハレル-ルプスキ症候群はさらなる課題を抱える可能性がありますが、多職種によるアプローチと適切なケアにより、患者の予後を改善し、より健康で長い生活を送る支援が可能となるでしょう。

もっと知りたい方へ
【写真あり・英語】ユニーク(Unique)による17p重複に関する情報シート
【写真あり・英語】ユアン-ハレル-ルプスキ症候群 – 米国国立医学図書館 MedlinePlus – 米国政府公式サイト – 最終更新 2018年10月1日
引用文献
- Yuan, B., Harel, T., Gu, S., Liu, P., Burglen, L., Chantot-Bastaraud, S., Gelowani, V., Beck, C. R., Carvalho, C. M., Cheung, S. W., Coe, A., Malan, V., Munnich, A., Magoulas, P. L., Potocki, L., & Lupski, J. R. (2015). Nonrecurrent 17p11.2p12 Rearrangement Events that Result in Two Concomitant Genomic Disorders: The PMP22-RAI1 Contiguous Gene Duplication Syndrome. American journal of human genetics, 97(5), 691–707. https://doi.org/10.1016/j.ajhg.2015.10.003
- Doco-Fenzy, M., Holder-Espinasse, M., Bieth, E., Magdelaine, C., Vincent, M. C., Khoury, M., Andrieux, J., Zhang, F., Lupski, J. R., Klink, R., Schneider, A., Goze-Martineau, O., Cuisset, J. M., Vallee, L., Manouvrier-Hanu, S., Gaillard, D., & de Martinville, B. (2008). The clinical spectrum associated with a chromosome 17 short arm proximal duplication (dup 17p11.2) in three patients. American journal of medical genetics. Part A, 146A(7), 917–924. https://doi.org/10.1002/ajmg.a.32195
- Shaw, C. J., Stankiewicz, P., Christodoulou, J., Smith, E., Jones, K., & Lupski, J. R. (2004). A girl with duplication 17p10-p12 associated with a dicentric chromosome. American journal of medical genetics. Part A, 124A(2), 173–178. https://doi.org/10.1002/ajmg.a.20355
- Franciskovich, R., Soler-Alfonso, C., Neira-Fresneda, J., Lupski, J. R., McCann-Crosby, B., & Potocki, L. (2020). Short stature and growth hormone deficiency in a subset of patients with Potocki-Lupski syndrome: Expanding the phenotype of PTLS. American journal of medical genetics. Part A, 182(9), 2077–2084. https://doi.org/10.1002/ajmg.a.61741
- Yuan-harel-lupski syndrome: Medlineplus genetics. (Last updated 2018 October). Retrieved January 6, 2025, from https://medlineplus.gov/genetics/condition/yuan-harel-lupski-syndrome/
- Perez, G., Barber, G. P., Benet-Pages, A., Casper, J., Clawson, H., Diekhans, M., Fischer, C., Gonzalez, J. N., Hinrichs, A. S., Lee, C. M., Nassar, L. R., Raney, B. J., Speir, M. L., van Baren, M. J., Vaske, C. J., Haussler, D., Kent, W. J., & Haeussler, M. (2024). The UCSC Genome Browser database: 2025 update. Nucleic Acids Research, gkae974. https://doi.org/10.1093/nar/gkae974
- Harrison, P. W., Amode, M. R., Austine-Orimoloye, O., Azov, A. G., Barba, M., Barnes, I., Becker, A., Bennett, R., Berry, A., Bhai, J., Bhurji, S. K., Boddu, S., Branco Lins, P. R., Brooks, L., Budhanuru Ramaraju, S., Campbell, L. I., Carbajo Martinez, M., Charkhchi, M., Chougule, K., … Yates, A. D. (2024). Ensembl 2024. Nucleic Acids Research, 52(D1), D891–D899. https://doi.org/10.1093/nar/gkad1049
ユアン-ハレル-ルプスキ症候群は、17番染色体の一部が重複することによって発生する稀な神経疾患です。ポトツキ-ルプスキ症候群やシャルコー-マリー-トゥース病の特徴を併せ持ち、発達遅延、低筋緊張、顔の特徴、末梢神経障害など多岐にわたる症状が見られます。診断と管理について解説します。
NIPT(新型出生前診断)について詳しく見る













