
 JA
JA
 EN
EN
 中文
中文





ルービンシュタイン・テイビー症候群1型(RSTS1)は、知的障害や成長障害、特徴的な顔の特徴を伴う稀な遺伝性疾患です。この症候群の診断方法、症状、管理方法、早期介入の重要性について解説します。

ダウン症の検査
気になる費用はこちら
この記事のまとめ
ルービンシュタイン・テイビー症候群1型(RSTS1)は、発達遅延、知的障害、特徴的な顔の特徴を持つ稀な遺伝性疾患です。この疾患は、主にCREBBP遺伝子の変異によって引き起こされ、症状は個々の患者によって異なります。早期診断と介入が重要で、適切なサポートにより充実した生活を送ることが可能です。
疾患概要


ルビンシュタイン・テイビー症候群(RSTS)は、先天的な異常、知的障害、そして特徴的な身体的特徴を伴う稀な遺伝性疾患です。この疾患は、おおよそ10万人から12万5千人に1人の割合で発症します。RSTSは、特に顔の特徴に関連しており、例えば、非常にアーチ状に盛り上がった眉毛、長いまつげ、下向きのまぶたの裂け目、広い鼻筋、そして鳥のくちばしのような鼻が見られます。その他の典型的な特徴としては、小さな頭(小頭症)、広い親指と大きな足の親指(拇指と拇趾)、高いアーチ状の口蓋、軽度の小顎症(小さな顎)、異常な笑顔やしかめっ面があります。
ルビンシュタイン・テイビー症候群の患者は、一般的に出生後の成長不足が見られ、これが原因で低身長になることが多いです。また、知的障害もよく見られます。さらに、腫瘍が形成されるリスクが高いとされ、医療的な監視が重要です。
この症候群は2つのタイプに分類されます。より一般的なタイプであるルビンシュタイン・テイビー症候群1(RSTS1)は、全体の約50~70%を占めます。このタイプは、主に16番染色体のp13.3領域にあるCREBBP遺伝子の変異によって引き起こされます。ルビンシュタイン・テイビー症候群2(RSTS2)は全体の約3%で、主に22番染色体のq13領域にあるEP300遺伝子の変異によって引き起こされます。
さらに重度の型である16p13.3染色体欠失症候群は、CREBBP遺伝子とその周辺の他の遺伝子を含む連続的な遺伝子欠失によって発症します。この記事では、主にRSTS1に焦点を当てています。
病因と診断の方法
ルビンシュタイン・テイビー症候群1型(RSTS1)は、主に16番染色体の16p13.3領域にあるCREB結合タンパク質(CREBBP)遺伝子の変異によって引き起こされます。この遺伝子は、細胞内で遺伝子の発現やクロマチン構造、細胞のさまざまな信号に対する反応を調節する重要な役割を果たします。具体的には、ヒストン(特にヒストンH3)の特定の部位でアセチル化を行い、クロマチンを転写活性化のための印を付けます。また、他のタンパク質と結びついて遺伝子発現を強化する役割もあります。さらに、CREBBPはDNA修復やタンパク質の安定性に関与する非ヒストンタンパク質のアセチル化も行い、細胞の正常な機能にとって欠かせません。
RSTS1のほとんどのケースは、デ・ノボ(新たに発生した)変異によって起こります。これは、遺伝子の変異が両親から受け継がれることなく自発的に発生することを意味します。このようなケースは全体の約99%を占めます。縦の遺伝(親から子への遺伝)は非常に稀ですが、いくつかの報告例があります。
この症候群は当初、染色体の異常と関連があると考えられており、初期の研究では染色体の一部が欠失する「マイクロデリーション」の可能性が示唆されていました。初期の遺伝子検査では重大な染色体異常が検出されなかったものの、後の研究により、CREBBP遺伝子に影響を与えるマイクロデリーションがこの病気の原因である可能性が高いことが確認されました。
そのため、RSTS1は通常、CREBBP遺伝子の片方のコピーに変異が生じることで発症し、ほとんどの症例は新たに発生した変異によるものですが、家族内で遺伝する場合もあります。これらの発見は、RSTS1の診断において遺伝子検査や家族歴の重要性を示しています。
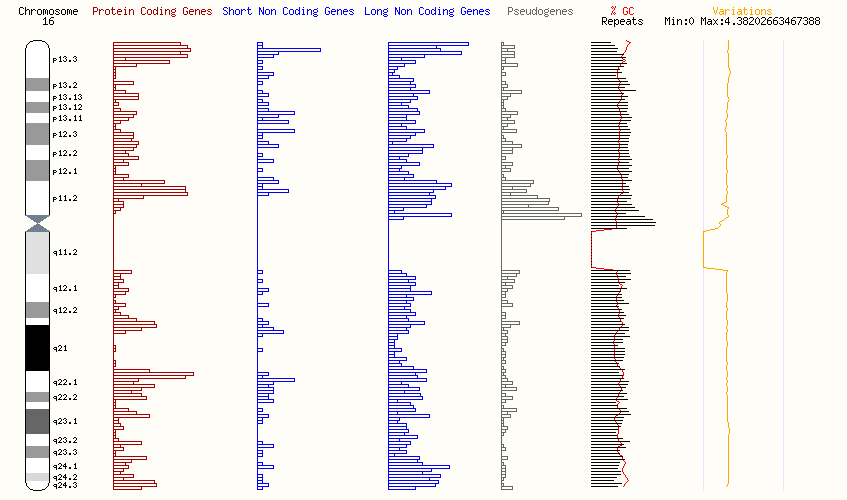
ルービンシュタイン・テイビ症候群1型(RSTS1)の診断は、主に臨床検査に基づいて行われます。約65%の症例では、細胞遺伝学的または分子遺伝的異常が検出され、診断が確認されます。しかし、RSTS1はサエスレ・ショッツェン症候群、フローティング・ハーバー症候群、コーネリア・デ・ランゲ症候群など、類似の症候群と特徴が重なることが多いため、診断を区別することが難しいことがあります。
もし、影響を受けた子どもに細胞遺伝学的または分子遺伝的異常が認められた場合、将来の妊娠において絨毛検査(胎盤の一部を採取して行う検査)による信頼できる出生前診断が可能です。しかし、出生前超音波検査ではRSTS1の診断はほとんどの場合、確定することができません。

RSTS1のほとんどの症例は偶発的であり、新たに発生した遺伝子変異(新規変異)が原因で、親から遺伝するものではありません。影響を受けた子どもがいる家庭では、研究により、今後の妊娠における再発リスクは約0.5%であると示唆されています。また、影響を受けた個人が子どもを持つことができる場合、遺伝の法則に従い、再発リスクは50%になります。この症候群は常染色体優性遺伝のパターンに従います。
診断方法として、FISH(蛍光 in situ ハイブリダイゼーション)やマイクロサテライト分析が有効であり、診断を確認するために役立ちます。早期の診断は、タイムリーなサポートを提供し、症候群を持つ個人の長期的な生活の質を改善するために重要です。
DNAシーケンシング技術の進展により、非侵襲的出生前検査(NIPT)は、出生前FISHや羊水検査といった侵襲的な方法に代わる、ますます信頼性の高い安全なスクリーニング方法として注目されています。NIPTは、母親の血液中に含まれる胎児の細胞を分析することで、母体や胎児の健康にリスクを与えることなく、正確な結果を提供します。このため、安全なスクリーニング方法を求める親にとって、ますます人気のある選択肢となっています。
疾患の症状と管理方法
ルービンシュタイン・テイビー症候群1型(RSTS1)は、重症度に差があるさまざまな臨床的特徴を持つ稀な遺伝性疾患です。主な特徴として、知的障害、短身、広い親指や大きな足の指(大趾)などが挙げられます。影響を受けた人々は、通常、高い弓形の眉毛、長いまつげ、下向きの目の開き(眼裂)、広い鼻梁、くちばし型の鼻、そして高い裂け目のような口蓋(上顎の屋根)といった顔の特徴を持っています。他にも、小さな顎(顎小症)や異常な笑顔やしかめ面が見られることがあります。さらに、広い大趾や指に残る胎児期の皮膚の隆起、ショール陰嚢(陰嚢が大きく覆われたような形)が一般的に見られます。
乳児期には、RSTS1は重篤な健康問題を引き起こすことがあります。例えば、16番染色体のp13.3領域に大きな欠失がある場合、脾臓の追加(副脾)、左心室の発育不全、異常な肺葉の分け方などの生命を脅かす奇形が見られることがあります。これらの重症例では、感染症や成長不良(成長障害)が発生し、場合によっては早期に命を落とすこともあります。

RSTS1は、いくつかの健康合併症とも関連しています。心血管の問題は一般的で、心房中隔欠損症や心室中隔欠損症、動脈管開存症、肺動脈狭窄などの先天性心疾患が見られます。これらの心疾患は治療が必要となることがあり、また、呼吸器系の問題、例えば喉頭の構造的異常や肺葉の異常によって呼吸困難を引き起こすことがあります。
この症候群は、良性および悪性の腫瘍が発生しやすくなることもあります。RSTS1患者は脳腫瘍、横紋筋肉腫、神経芽腫、白血病など、さまざまな腫瘍のリスクが高くなります。この腫瘍リスクの増加は、RSTS1患者の約5%に見られます。
整形外科的な問題もよく見られます。膝蓋骨の脱臼、関節の緩み、先天的な脱臼などが一般的です。これらの関節の異常は、移動に影響を与え、手術治療が必要となることがあります。その他の筋骨格系の問題には、脊柱側弯症(側弯症)、脊柱後弯症(後弯症)、反復する骨折などがあります。

RSTS1では、歯や口の健康問題も頻繁に見られます。歯の数の異常、エナメル質の低形成、歯の変色、交差咬合などが多くの患者で見られます。胃食道逆流症も一般的で、これが歯のエナメル質の摩耗に影響を与えることがあります。
行動面では、RSTS1の人々は注意力、社会的相互作用、運動発達において課題を抱えることがよくあります。注意力が続かない、社会的な障害、表現言語の発達の遅れなど、いわゆる自閉症様の行動がよく見られます。これらの発達の遅れに対処するためには、行動療法や特別支援教育が必要となることが多いです。
また、RSTS1の患者は、便秘などの消化器系の問題や、先天性の異常(例えば、隠れ精巣や発育不良の腎臓)などの泌尿生殖器系の異常もよく見られます。摂食に関する問題や嚥下困難も見られ、これがケアをさらに複雑にすることがあります。

RSTS1は多面的な疾患であり、患者は身体的、発達的、行動的なさまざまな課題に対処するために、包括的なケアが必要です。早期診断と介入は、適切なサポートを提供し、長期的な成果を改善するために非常に重要です。
ルービンシュタイン・テイビー症候群1型(RSTS1)の管理は、個々の症状やニーズに対応することを中心に行われ、しばしば専門医のチームによるアプローチが必要です。この疾患は健康の複数の側面に影響を及ぼすため、包括的なケアが求められます。小児科医、心臓病専門医、整形外科医、耳鼻科医、泌尿器科医、腎臓科医、歯科専門医、理学療法士、言語病理学者、栄養士などが関わり、定期的なモニタリングとケアが最良の結果を得るために不可欠です。
乳児や幼児にとって、早期介入が重要です。作業療法、理学療法、言語療法、授乳療法、そして乳幼児精神保健サービスなどを提供する早期介入プログラムは、個々のニーズに合わせて行われます。アメリカでは、これらのサービスは家庭で提供されることが多く、子供の発達を支援します。3歳から5歳の子供には、発達支援のために地域の公立学校区の発達型保育園や個別教育計画(IEP)を通じた支援が推奨されます。

発達小児科医との継続的な相談も重要で、適切な地域・州・教育機関の支援を受けることができます。子供が思春期に入ると、成人に向けた移行計画をIEPに組み込むことが望ましいです。家族や学校は、課題に対応するために、時間の柔軟性や座席の変更などの調整が必要になることがあります。
理学療法は、運動能力を高め、脊柱側弯症や関節拘縮、股関節脱臼などの整形外科的合併症のリスクを減らすために推奨されます。特に手指や足の細かな動きに影響を与える場合、作業療法も有効です。作業療法は、食事、身だしなみ、着替え、書き込みなど、日常生活に必要な細かな運動機能の改善に役立ちます。
授乳や飲み込みの問題については、定期的な評価が必要です。必要に応じて、言語療法士や作業療法士による授乳療法を通じて、協調や感覚的な問題に対応します。重度の授乳障害がある場合、胃管(NGチューブやGチューブ)の使用が検討されることもあります。

表現言語に困難がある場合、代替的なコミュニケーション手段(AAC)を評価することが推奨されます。AACには、ピクチャーエクスチェンジシステムのような低技術なものから、音声生成デバイスのような高技術なものまであります。言語病理学者による評価を通じて、最適なコミュニケーション方法を選ぶことができます。
自閉症スペクトラム障害の治療に使用される応用行動分析(ABA)などの療法が有効です。ABAは、子供の行動、社会性、適応力を個別にサポートし、発達を促進します。注意欠陥・多動性障害(ADHD)の薬物治療も、注意力や気分の管理に役立つことがあります。
攻撃的または破壊的な行動が見られる場合は、小児精神科医との相談が必要になることがあります。しかし、影響を受けた子供は一般的に「親しみやすく、幸せで、おおらか」と表現されます。
症状の進行、治療への反応、新たな症状の出現を監視するために、定期的な評価が不可欠です。これには、RSTS1専用の成長曲線で成長を追跡し、毎年目や聴力の検査を行い、心臓、歯科、腎臓の問題を定期的にチェックすることが含まれます。呼吸や授乳に関する問題には、早期介入が重要です。

RSTS1は多面的な疾患であるため、心臓、骨格、聴覚、泌尿器系の専門医を含むチームによるケアが最適です。理学療法、外科手術、その他の治療方法は、脊柱側弯症などの骨格異常や関節脱臼などの整形外科的問題に対応するために必要です。親指や足の親指に問題がある場合、靴を履くことが難しくなるため、手術が検討されることもあります。
RSTS1の影響を受けた個人やその家族には、遺伝カウンセリングが推奨されます。遺伝カウンセリングは、遺伝的な側面を理解し、再発のリスクを話し合うために役立ちます。特に、影響を受けた個人が子供を持つことを考えている場合は重要です。

思春期に入ると、肥満、気分障害、行動の変化など、年齢に関連する問題に対応するために、管理方法を調整する必要があります。独立性を維持し、適切な医療を受け、社会的・感情的なサポートを提供することが重要です。
早期診断と個別化された介入により、多くのRSTS1患者は適切な医療、教育、行動療法のサポートを受けながら充実した生活を送ることができます。包括的なケア、継続的なモニタリング、個別の治療が長期的な結果を改善するために重要です。
DNAシーケンシング技術の進歩により、非侵襲的な出生前診断(NIPT)が信頼性の高い安全なスクリーニング方法として注目されています。NIPTは母体の血液中に含まれる胎児細胞を分析し、母子の健康にリスクを与えることなく正確な結果を提供します。これにより、妊娠中の安全なスクリーニング方法を求める親にとって人気の選択肢となっています。
将来の見通し
ルービンシュタイン・テイビー症候群1型(RSTS1)に関連する障害を持つ90%以上の方々が成人期を迎えており、その中には67歳まで生存した方も報告されています。これは、RSTS1の方々が成人期まで生きることが可能であることを示しています。ただし、これらの患者の医療管理は個別の支援が必要で、標準的なガイドラインが少ないため、時間がかかることが多いです。また、多くの成人は高度な遺伝子検査を受けていないため、十分に認識されていないこともあります。
RSTS1に影響を受けた方々が直面する課題は確かにありますが、適切なケアと支援があれば、RSTS1の子どもたちも充実した人生を送ることができます。適切な医療、教育、行動療法を通じて、その可能性は大いに広がり、豊かな未来を切り開くことができるのです。

もっと知りたい方へ
【支援団・日本】ルビンシュタイン・テイビ症候群児親の会 こすもす公式ブログ
【写真あり・英語】RTS Support Group UK | Aaraの証 | Ambaの証 | Jackの証
引用文献
- Hennekam, R. C. M. (2006). Rubinstein–Taybi syndrome. European Journal of Human Genetics, 14(9), 981–985. https://doi.org/10.1038/sj.ejhg.5201594
- Milani, D., Manzoni, F. M., Pezzani, L., Ajmone, P., Gervasini, C., Menni, F., & Esposito, S. (2015). Rubinstein-Taybi syndrome: clinical features, genetic basis, diagnosis, and management. Italian journal of pediatrics, 41, 4. https://doi.org/10.1186/s13052-015-0110-1
- Rubinstein-taybi syndrome—Symptoms, causes, treatment | nord. (Last updated July 2023). Retrieved January 15, 2025, from https://rarediseases.org/rare-diseases/rubinstein-taybi-syndrome/
- Stevens CA. Rubinstein-Taybi Syndrome. 2002 Aug 30 [Updated 2023 Nov 9]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1526/
- Orphanet. (Last updated August 2019). Rubinstein-Taybi syndrome. Reviewed by Dr L.A. [Leonie] MENKE. Retrieved from https://www.orpha.net/en/disease/detail/783
- Online Mendelian Inheritance in Man. RUBINSTEIN-TAYBI SYNDROME 1; RSTS1. (Last updated February 2015 by Cassandra L. Kniffin). Retreived from https://omim.org/entry/180849
- Online Mendelian Inheritance in Man. CHROMOSOME 16p13.3 DELETION SYNDROME, PROXIMAL. (Last updated February 2015 by Cassandra L. Kniffin). Retreived from https://omim.org/entry/610543
- Perez, G., Barber, G. P., Benet-Pages, A., Casper, J., Clawson, H., Diekhans, M., Fischer, C., Gonzalez, J. N., Hinrichs, A. S., Lee, C. M., Nassar, L. R., Raney, B. J., Speir, M. L., van Baren, M. J., Vaske, C. J., Haussler, D., Kent, W. J., & Haeussler, M. (2024). The UCSC Genome Browser database: 2025 update. Nucleic Acids Research, gkae974. https://doi.org/10.1093/nar/gkae974
- Harrison, P. W., Amode, M. R., Austine-Orimoloye, O., Azov, A. G., Barba, M., Barnes, I., Becker, A., Bennett, R., Berry, A., Bhai, J., Bhurji, S. K., Boddu, S., Branco Lins, P. R., Brooks, L., Budhanuru Ramaraju, S., Campbell, L. I., Carbajo Martinez, M., Charkhchi, M., Chougule, K., … Yates, A. D. (2024). Ensembl 2024. Nucleic Acids Research, 52(D1), D891–D899. https://doi.org/10.1093/nar/gkad1049
ルービンシュタイン・テイビー症候群1型(RSTS1)は、知的障害や成長障害、特徴的な顔の特徴を伴う稀な遺伝性疾患です。この症候群の診断方法、症状、管理方法、早期介入の重要性について解説します。
NIPT(新型出生前診断)について詳しく見る













