 JA
JA
 EN
EN
 中文
中文



Erin E. Salo-Mullen, MS, MPH, CGC, Patricio B. Lynn, MD, Lu Wang, MD, PhD, Michael Walsh, MD,Anuradha Gopalan, MD, Jinru Shia, MD,Christina Tran, BS, Fung Ying Man, MD, MSc,Sean McBride, MD, MPH, Mark Schattner, MD,Liying Zhang, MD, PhD,Martin R. Weiser, MD,and Zsofia K. Stadler, MD
医学部、スローンケタリング記念がんセンター、1275ヨーク平均、ニューヨーク、ニューヨーク、10065
手術、スローンケタリング記念がんセンター、1275ヨーク平均、ニューヨーク、ニューヨーク、10065
病理学、スローンケタリング記念がんセンター、1275ヨーク平均、ニューヨーク、ニューヨーク、10065
Radiation Oncology, Memorial Sloan Kettering Cancer Center, 1275 York Ave., New York, NY 10065
Weill Cornell Medical College, 1300 York Ave, New York, NY 10065 医学部 手術、Weill Cornell Medical College, 1300 York Ave, New York, NY 10065
要約
リンチ症候群は、DNAミスマッチ修復(MMR)遺伝子における病原性突然変異によって引き起こされる常染色体優性疾患である。 知的障害や先天異常などの臨床的特徴と一般的に関連しているが、隣接する遺伝子欠失もまた、がん素因症候群を引き起こすことがある。染色体2p16.3‐p21の欠失により引き起こされたリンチ症候群の52歳男性について報告する。 患者は知的障害を有し、MSH2およびMSH6蛋白発現の欠如を伴うMMR欠損を示す偶発的に同定された同時性S状結腸腺癌を伴う前立腺腺腺癌を呈した。 家族歴は明らかではなかった。 身体診察では、低身長、短い額と短い親指を伴う小脳症、手の短指症、手掌の横方向のしわ、足底の色素沈着過剰を伴う広く小さな足が明らかになった。 患者は、pT3N1S状腺癌のために回腸直腸吻合を伴う結腸全摘術を受けた。 MSH2、MSH6、およびEPCAM遺伝子の生殖細胞系遺伝子検査により、完全な遺伝子欠失が明らかになった。 SNPアレイに基づくDNAコピー数分析は、2p16.3‐p21で4.8Mbの欠失を同定した。 3つのリンチ症候群関連遺伝子に加えて、欠失した染色体切片は、NRXN1、CRIPT、CALM2、FBXO11、LHCGR、MCFD2、TTC7A、EPAS1、PRKCE、および15の遺伝子を包含した。 家族性大腸腺腫症などの他の遺伝性がん素因症候群では、先天性遺伝子欠損が報告されている。 著者らの報告および文献のレビューは、2p16‐p21染色体領域内の隣接遺伝子欠失がリンチ症候群の稀な原因であるが、異なる表現型特徴を呈することを示唆し、この症候群実体の認識および認識の必要性を強調する。
共著者: Zsofia K. Stadler, MD, 1275 York Ave. Box 295, New York, NY 10065, stadlerz@mskcc.org, Telephone:
646-888-4039、FAX:646-888-4051
*共著者
利益相反:無
キーワード
リンチ症候群;染色体欠失;前立腺がん、大腸がん、 2p
はじめに
リンチ症候群は、DNAミスマッチ修復(MMR)遺伝子(MLH1、MSH2、MSH6、PMS2)の病原性突然変異およびEPCAM(TACSTD1)遺伝子の3’エキソンの欠失によって引き起こされる常染色体優性の疾患であり、大腸癌、子宮内膜癌、卵巣癌、胃癌、上部尿路癌のリスク増加と関連している[1]。 MSH2、MSH6、およびEPCAM遺伝子は、染色体2p上に位置する。 リンチ症候群の診断過程には、MMR蛋白発現に対する腫瘍免疫組織化学(IHC)染色、マイクロサテライト不安定性(MSI)分析、および生殖細胞系遺伝子検査がある[1]。
隣接遺伝子欠失症候群は、特定の染色体断片から複数の隣接遺伝子が欠失した結果である。 知的障害、発達遅延、先天異常などの様々な臨床的特徴を有する患者では、隣接する遺伝子欠失が同定されている[2]。 多くの隣接する遺伝子欠失は、関連する表現型がしばしば複雑で、生殖適応度の低下につながるため、de novo事象の結果である[2]。
以前に報告されているが、隣接する遺伝子欠失はがん素因症候群のまれな原因である[3]。 本稿では、MSH2、MSH6、およびEPCAM遺伝子を包含する染色体2p16.3‐p21における隣接遺伝子欠失により引き起こされたリンチ症候群患者について報告する。 過去の報告のレビューと共に、我々は、発達異常の根底にある原因として隣接する遺伝子欠失症候群を認識する必要性、および年齢に応じて罹患者のがんリスクおよび管理を考慮する必要性を強調する。
方法
臨床像
病因不明の知的障害の既往のある52歳男性が、22ng/mLのPSAスクリーニング後の生検でグリソン7前立腺腺癌と診断された。 骨スキャンは陰性であった。 外照射療法を予定していたが、放射線治療中の残存を遵守できなかったため実施しなかった。 手術管理のため当院を受診した際、患者の介護者から直腸あたり1か月間の明赤色血液の病歴が報告された。 術前評価全身および前立腺磁気共鳴画像法(MRI)は、原発腫瘍および限局性前立腺疾患が疑われる遠位S状結腸の同心性肥厚を明らかにした。 軟性S状結腸鏡検査では、S状結腸に部分的に周囲の非閉塞性の大きな腫瘤が同定され、生検では浸潤性低分化癌が確認された。 MMR蛋白質のIHC染色はMSH6の欠如を示した。 胸部、腹部および骨盤の病期分類CTでは、遠隔転移の証拠は示されなかった。
MMR IHCの異常結果に基づき、患者はClinical Genetics Serviceに紹介された。 4世代家系図では、がん、大腸ポリープ、および発達障害については明らかにされていなかった。 患者さんには子供はいません。 兄弟3人、姉妹2人が40~50代で生きている。 お母さんとお父さんは70代で生きていて元気だ。 同情は否定された。 家族はアフリカ系アメリカ人の家系である。 知的障害出生。 言語理解は単純なコマンドに限定され、言語表現は少数の単語に限定される。 彼は両親と一緒に暮らし、すべてのセルフケア活動に助けを求めている。 彼は毎日の成人向けプログラムに参加し、日々の監督と支援を必要としている。 身体診察では、低身長(4’8′)、短い額と短い親指を伴う小脳症、短指症、手掌横ひだ、指先の肉質のパッド、足底の色素沈着過剰を伴う広く小さな足が明らかになった(図1a)。 文書によるインフォームド・コンセントを患者の親/保護者から取得し、写真を公開に使用した。
遺伝学的評価
結腸腫瘍におけるMMR欠損の存在に基づき、リンチ症候群の可能性が高まった。 リンチ症候群と前立腺がんとの関連の可能性も指摘された。 患者の母親は、MSH6、MSH2、およびEPCAM遺伝子の生殖細胞系遺伝子検査についてインフォームド・コンセントを提供し、生殖細胞系DNA標本を得た。 MSH6、MSH2、およびEPCAM遺伝子の生殖細胞系遺伝子検査をMSK Diagnostic Molecular Genetics Laboratory(配列決定および多重ライゲーション依存性プローブ増幅)で実施した。 最初の検査結果に続いて、患者の母親はMSK Clinical Cytogenetics Laboratoryで実施されたSNPアレイに基づくDNAコピー数分析(Affymetrix CytoScan HD Assay)に対する追加のインフォームドコンセントを提供した。
遺伝子検査の結果
遺伝子検査により、MSH6、MSH2、およびEPCAM遺伝子のヘテロ接合の完全遺伝子欠失が明らかになった。 この知見に基づき、SNPアレイに基づくDNAコピー数分析を行い、2p16.3‐p21で4.8Mbの欠失を同定した。 3つのリンチ症候群関連遺伝子に加えて、欠失した染色体セクションには以下の24個の遺伝子が含まれる: PRKCE、EPAS1、TMEMAS247、ATPV1E2、HORQ、PIGF、CRIPT、SOCS5、LOC38948、LOC100134259、MCFD2、TTC7A、C2orf61、CLM2、MIR559、KCNK12、FBXO11、FOXN2、PPP1R21、STON1、GTF2A1L、LHCGR、FSHR、およびNRXN1(図1b)。
臨床シークエラ
MSH6、MSH2、およびEPCAM遺伝子の完全な遺伝子欠失の最初の所見に基づき、リンチ症候群の診断を下した。 異時性大腸がんのリスクが高く、大腸内視鏡検査のための腸の準備を遵守することが困難であることを考慮して、回腸直腸吻合術を伴う結腸全摘術の選択肢が議論され、様々な外科的アプローチのリスクと利益を考慮した後、家族に受け入れられた。
直腸部分切除術および回腸中部直腸吻合術を伴うロボット式大腸全摘術を受けた。 外科的病理学的検査により、リンパ管浸潤を伴うpT3N1M0低分化型直腸S状結腸腺癌が確認され、腫瘍浸潤リンパ球の増加が認められた;42個のリンパ節のうち2個が疾患陽性であった。 粘膜内癌を伴う別個の管状絨毛腺腫も認められた。 IHC染色では、MSH2およびMSH6タンパク質の発現は認められなかったが、MLH1およびPMS2タンパク質の発現は正常であった(図2a-d)。 患者は順調な外科的回復を示し、カペシタビンおよびオキサリプラチン(CAPEOX)を含む補助化学療法を受けた。 化学療法は、血小板減少症および貧血を含む累積副作用のため、早期に中止された。
患者は最終的に、神経温存両側リンパ節郭清を伴う恥骨後根治的前立腺摘除術を受けた。 病理学的には、5個の良性リンパ節を有するpT3a多中心性グリソン7(3+4)浸潤性腺癌が明らかになった。 IHC染色では、MSH2およびMSH6蛋白質発現は認められなかったが、MLH1およびPMS2蛋白質の発現は正常であった(図2e-h)。
経過観察の臨床遺伝カウンセリングでは、隣接する遺伝子欠失が患者の知的障害とリンチ症候群の両方の原因であると推定されることが議論された。 さらに、大腸がんおよび前立腺がんのいずれにおいてもMSH2およびMSH6タンパク質発現が認められないことを考えると、いずれのがんもリンチ症候群に起因する。 同定された隣接遺伝子欠失はおそらく新規起源であるが、患者の第一度近親者には個別化された遺伝的リスク評価が推奨された。
考察
家族性大腸腺腫症[4, 5]などの遺伝性がん素因症候群の状況では、先に隣接する遺伝子欠失が報告されている。 著者らの報告は、知的障害の既往があり、隣接遺伝子欠失の結果リンチ症候群を有することが判明した新たな同時性癌診断を有する成人男性について述べる。 文献のレビューから、2p16p21を包含する類似の欠失を有する少数の個体が既に記載されている。[6、7、8、9、10](図1c、表1) 著者らの患者の身体的特徴および知的障害のレベルは、2p16.2p21欠失を有する個人の過去の報告と類似しているが、患者は記述されるべき最も古い罹患個人である。
4.8 Mbの欠失に含まれる遺伝子をレビューしたところ、多くの遺伝子に潜在的な医学的意義が認められた(表2)。 EPAS1、MCFD2、およびCALM2遺伝子の突然変異は、それぞれ、赤血球増加症ファミリー4型、第V因子および第VIII因子複合欠損症、およびLong-QT症候群と関連することが報告されている。 著者らの患者は、エリスロポエチンレベルが正常である慢性腎疾患に起因する可能性が高い正色素性正球性貧血を示したが、赤血球増加家族性4型の証拠はなかった。 心電図はQT延長症候群を示さなかった。 われわれは、遺伝子型-表現型機序が、患者がこれらの状態を発現しない理由を説明する可能性があるという仮説を立てる。 赤血球増加家族性4型は、EPAS1(HGNC ID #3374)における有害な突然変異とは対照的に、活性化と関連している。 同様に、MCFD2およびCALM2遺伝子における記載された変化には、スプライス部位、フレームシフトおよびミスセンス突然変異が含まれているが、機能欠失は含まれていない(それぞれ、HGNC ID # 18451および1445)。2p16p21欠失を有する以前に記載された患者も発達遅延/知的障害を経験し、我々の患者において欠失することが見出されたCRIPTおよびNRXN1の少なくとも2つの遺伝子が中枢神経系欠損と関連していることを考慮すると、この患者の知的障害の病歴は同定された隣接遺伝子欠失に関連している可能性が高いと考えられる。 注目すべきことに、著者らの患者で同定された欠失は27遺伝子の欠失を生じたが、SIX3遺伝子は含まれなかった。 以前の報告のいくつかでは、SIX3は全前脳症と関連していることが指摘されている。
リンチ症候群に関して、本症例はまた、異時性大腸がんのリスクを考慮した外科的管理に関する情報に基づく意思決定の重要性を強調している。 Parryら MMR遺伝子変異を有する被験者50人のコホートにおいて、「広範な結腸切除術」(「回腸直腸吻合術を伴う結腸全摘術および回腸S状結腸吻合術を伴う結腸亜全摘術」と定義)後に異時性癌は認められなかったのに対し、区域切除術を受けた突然変異キャリア332人では異時性大腸癌は74人であった[11]。 この症例では、家族が結腸全摘術を進める決定には、将来のがんリスク、継続中の年1回の結腸鏡検査の必要性、生活の質に関する考慮が含まれていた。 本症例に例示される別の点は、前立腺がんとリンチ症候群との関連である。 相反するデータは存在するが、Raymondらの研究がある。 一般集団と比較して、リンチ症候群突然変異キャリアにおける前立腺がんの全ハザード比は1.99(95% CI、1.31~3.03; P = 0.0013)と算出された[12]。 実際、患者の前立腺がん組織におけるDNAミスマッチ修復蛋白質の免疫組織化学的染色は、リンチ症候群遺伝子欠失が患者の前立腺がん発生において役割を果たしていることを示唆するMSH2およびMSH6蛋白質の欠失を明らかにした。 最後に、患者の最初の直腸S状結腸腺癌生検ではMSH6蛋白のみが認められなかったが、生検標本で報告されたMSH2の保持は、エッジアーチファクトなどの技術的問題による可能性が高いと考えられる。 患者の直腸S状結腸腺癌切除標本のMMR IHC分析では、MSH2およびMSH6タンパク質の両方が認められず、患者が同定した遺伝子欠失を反映していた。
染色体2p16.3‐p21における隣接遺伝子欠失の著者らの報告は、以前に報告された症例と強い類似性を示した。 この患者の知的障害の病因の調査はこれまで行われていなかった。 より最近開発された技術は、患者が小児であったときには利用できなかったが、間質性染色体欠失を検出する能力をもたらした。 同定された隣接遺伝子欠失症候群は、患者の知的障害、形態異常の特徴、および癌の診断の可能性のある説明を提供している。 発達遅滞、知的障害、先天異常のある小児が先進的な遺伝子技術を用いて評価されるにつれて、がん素因遺伝子を含む染色体再構成のさらなる症例が同定される可能性が高い。 医療提供者、患者およびその介護者は、そのような診断の設定における関連するがんリスクを認識することが重要であり、それにより、適切な年齢で、集中的ながんサーベイランス、予防的処置、および化学予防のような適切な医学的管理を検討することができる。
謝辞
本研究は、Romeo Milio Lynch Syndrome Foundation、Kate and Robert Niehaus Clinical Genetics Initiative、およびNIH/NCI Cancer Center Support Grant P30 CA008748から一部資金提供を受けた。
参考文献
1.Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. リンチ症候群のレビュー:病歴、分子遺伝学、スクリーニング、鑑別診断、および医学的転帰。 クリン・ジェネット 2009; 76(1):1–18.
2.Nussbaum, RL., McInnes, RR., Williard, HF. ThompsonとThompson Genetics in Medicine。 6.Saunders/Elsevier; Philadelphia: 2004. p. 164-165.
3.Adams SA, Coppinger J, Saitta SC, Stroud T, Kandamurugu M, Fan Z, Ballif BC, Shaffer LG,Bejjani BA. 遺伝子型初回診断の影響:aCGHによる癌素因を伴う微小欠失および微小重複症候群の検出。 Genet Med. 2009; 11(5):314–322. [PubMed:19365269]
4.Herrera L, Kakati S, Gibas L, Pietrzak K, Sandberg AA. 5qの間質欠失のある男性におけるガードナー症候群 Am J Med Genet. 1986; 25(3):473–476. [PubMed: 3789010]
5.Raedle J, Friedl W, Engels H, Koenig R, Trojan J, Zeuzem S. A de novo de novo chromosome delletementation of chromosome q5が家族性大腸腺腫症、形態異常、軽度の精神遅滞を引き起こす。 Am JGastroenterol. 2001; 96(10):3016–3020. [PubMed: 11693343]
6.Petit P, Fryns JP. マーカー染色体形成を伴う間質欠失2pは欠失セグメントの形成を伴い、安定な無動原体マーカー染色体を生じる。 ジェネット・クーンズ 1997; 8(4):341-343.[PubMed: 9457505]
7.Sanders SR, Dawson AJ, Vust A, Hryshko M, Tomiuk M, Riordan D, Prasad C. 染色体2p16.2p21の間質性欠失。 クリンジスモルフォール。 2003; 12(3):183–185. [PubMed: 14564157]
8.Lucci-Cordisco E, Zollino M, Baglioni S, Mancuso I, Lecce R, Gurrieri F, Crucitti A, Papi L, NeriG, Genuardi M. MSH2遺伝子座の喪失を伴う新規微小欠失症候群および遺伝性非ポリポーシス大腸がん。 クリン・ジェネット 2004; 67(2):178–182.
9.Collins DL, Schimke RN. CHGアレイにより検出されたがん素因について。 Genet Med.2011; 13(11):982. [PubMed: 22051689]
10.Morton, SA (Adams), Coppinger, J., Ballif, BC., Shaffer, LG., Ellison, JW., Saitta, SC., Stroud, T.,Manickam, K., Fan, Z. Response to the letter by Collins and Schimke. Genet Med. 2011; 13(11):982–983. [PubMed: 22051689]
11.Parry S, Win AK, Parry B, Macrae FA, Gurrin LC, Church JM, Baron JA, Giles GG, Leggett BA,Winship I, Lipton L, Young GP, Young JP, Lodge CJ, Southey MC, Newcomb PA, Le Marchand L,Haile RW, Lindor NM, Gallinger S, Hopper JL, Jenkins MA. ミスマッチ修復遺伝子突然変異キャリアに対する異時性大腸がんのリスク:より広範な結腸手術の利点。 消化管2011; 60(7):950-957。 [PubMed: 21193451]
12.Raymond VM, Mukherjee B, Wang F, Huang SC, Stoffel EM, Kastrinos F, Syngal S, Cooney KA,Gruber SB. リンチ症候群の男性における前立腺がんリスクの上昇。 J Clin Oncol. 2013;31(14):1713–1718. [PubMed: 23530095]
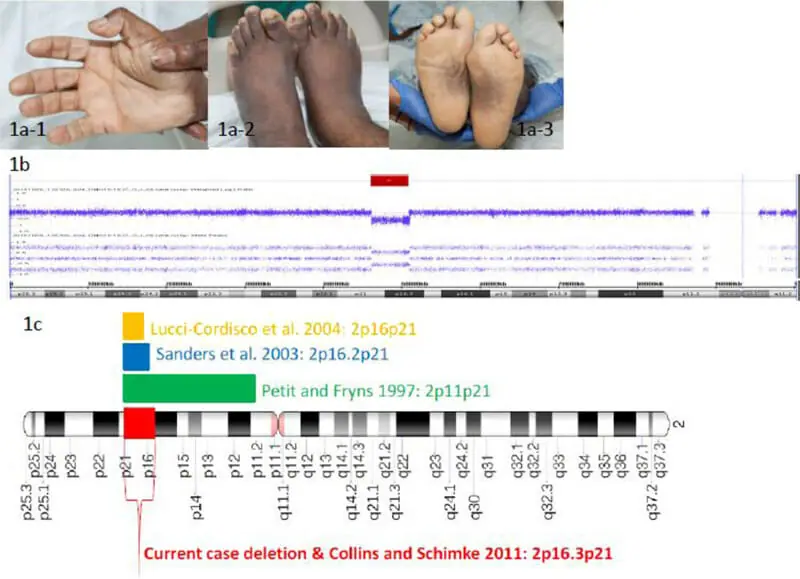
図1 1a)手指および足指の短指症(1a-1および1a-2)、手掌横ひだ(1a-1)、足底の色素沈着過剰を伴う広く小さな足(1a-3)
1b)染色体2のpアーム全体のDNAコピー数(log2比)および対立遺伝子ピークプロファイルを示す発端者の写真、赤い棒は2p16.3-p21での間質欠失(~4.8Mb)を示す。
1c)以前に報告された症例で同定された連続的な遺伝子欠失を示す染色体2のアニメーション
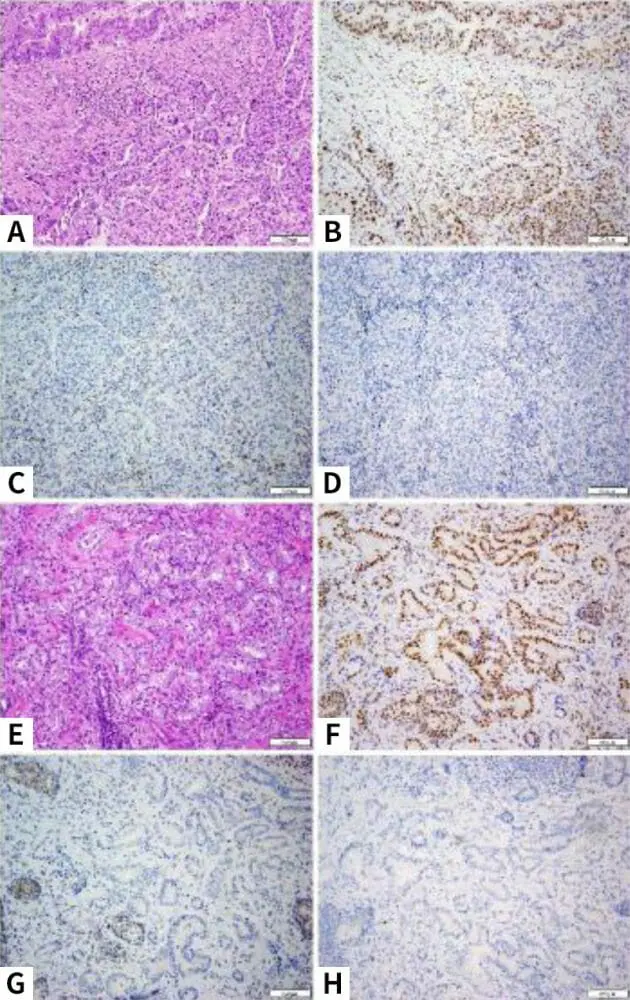
図2 患者の結腸直腸腺癌(a、H&E)は、腺形成成分(左上)と充実性成分を伴う混合した組織学的パターンを示す。 免疫組織化学検査では、腫瘍はPMS2(b)およびMLH1(図示せず)の核染色の存在を示すが、MSH2(c)およびMSH6(d)の染色は認められない。 患者の前立腺腺癌は組織学的に典型的な腺房単位(e,H&E)を示す。 免疫組織化学的検査では、この前立腺がんはまた、PMS2(f)およびMLH1(図示せず)の核染色の存在を示すが、MSH2(g)およびMSH6(h)の染色は認められない。
表1 2p16p21領域を含む欠失の報告例
| 参考 | 削除 | 表現型の説明 |
|---|---|---|
| Petit and Fryns 1997 | 2p11p21+アセフル | 軽度の異形および中等度の精神遅滞 |
| サンダースら 2003 | 2p16.2p21 | 心房中隔欠損、軽度の末梢機能低下、顕著な鼻橋、長い滑らかな好中球、軽度の発達遅延 |
| Lucci-Cordiscoら 2004 | 2p16p21 | 発達遅滞、思春期早発、低身長、額が狭い近接頭蓋骨、厚い眉毛を伴う大きな水平眼、鼻孔形成不全を伴う顕著なコルメラ、短母指、単純耳、多毛症、全身性肥満、短指症、両側第2~第3趾間の皮膚合指症、中等度の精神遅滞、結腸腺癌(37歳) |
| コリンズ・シンク2011 | 2p16.3p21 | 発達遅滞、精神遅滞、低身長、小頭症、下頭症、21歳時の本質的に正常な結腸鏡検査(炎症性偽ポリープのみ同定) |
| Mortonら 2011 | 17.6 Mb (SIX3, MSH2, and MSH6) | 全脳症を伴う胎児 |
| Mortonら 2011 | 192 kb(MSH6およびFBX011) | 発達遅延および形態異常の特徴 |
| Mortonら 2011 | 537 kb(MSH6およびFBX011) | 発達遅延および形態異常の特徴 |
| 現在の報告 | 2p16.3p21(4.8Mb) | 知的障害、低身長、額が狭く短い親指を伴う短頭症、短指症、足底の色素沈着過剰を伴う広く小さな足 |
表2 現在の患者に潜在的な医学的意義を有する特定の遺伝子
| 欠失遺伝子 | HGNC(HUGO遺伝子命名委員会)ID番号 | 関連する表現型 | 現在の患者で発現している表現型 |
|---|---|---|---|
| EPAS1 | 3374 | 赤血球増加家族性4型(通常は活性化突然変異と関連) | なし |
| CRIPT | 14312 | 出生後発育不全、小頭症、顔面異形、眼の異常、発達遅滞、再発性感染症、中枢神経系欠損 | 出生後発育不全、異形顔貌、眼異常、発達遅滞、中枢神経系欠損 |
| MCFD2 | 18451 | 第V因子および第VIII因子複合体欠損症 | なし |
| TTC7A | 19750 | 早期発症の炎症性腸疾患および腸閉塞 | なし |
| CALM2 | 1445 | QT延長症候群 | なし |
| EPCAM、MSH2、MSH6 | 1152973257329 | リンチ症候群 | リンチ症候群結腸がんおよび前立腺がん |
| LHCGR | 6585 | 男性の二次性的発達障害 | 弊社サービスでは評価しておりません。 |
| NRXN1 | 8008 | ピット・ホプキンス様症候群2 | 知的障害、精神運動面での遅れ、言葉の制限、コミュニケーションや社会性の障害、細い眉、突出した鼻、広い口、太い耳、笑顔や笑い声、手を振る動作が多く、幸せで興奮しやすい態度、斜視、低身長、小さな手と足、扁平足、手足の指先に異常な肉付きがあること |


