 JA
JA
 EN
EN
 中文
中文



Michelle Dolan, MD1, Nancy J. Mendelsohn, MD2,3, Mary Ella Pierpont, MD, PhD2,3,4, Lisa A. Schimmenti, MD4, Susan A. Berry, MD4, and Betsy Hirsch, PhD1
目的
アレイベースの比較ゲノムハイブリダイゼーションによる全ゲノム解析は、新規の微小欠失および/または微小重複症候群の発見の急速な増加をもたらした。 ここでは、19p13.13の新規微小欠失/微小重複の臨床的および細胞ゲノム相関について述べる。 方法:アレイベース比較ゲノムハイブリダイゼーション分析のために細胞遺伝学研究所に紹介された患者の中で、19p13.13内に欠失を有する4人と重複を有する1人を同定した。 確認的蛍光in situハイブリダイゼーションおよび親の検査を実施した。 詳細な臨床所見およびアレイプロフィールをレビューし、比較した。
結果:19p13.13の欠失を有する患者は、独特の一連の表現型異常を共有する。 発達障害に加えて、微小欠失は、過成長、巨頭症、および眼科的および消化管所見がみられた。対照的に、単一の微小重複は、成長遅延および小頭症がみられた。 結論:この領域のコピー数変異に関連する一貫した臨床所見を集積すると、19p13.13微小欠失/微小重複症候群と呼ぶのが妥当であろう。 16個の遺伝子を含むオーバーラップの約311~340 Kbの最小領域が同定された。 候補遺伝子としては、MAST1、NFIX、およびCALRが挙げられる。 この症候群の同定は、このコピー数変異型を有する患者に診断的精密検査とフォローアップをすることを推奨する。 詳細な臨床データとアレイデータの統合は、患者ケアとヒトゲノム研究の両方を進歩させるために重要である。
Genet 2010:12(8):503-511を満たす。
キーワード:
原因不明の発達障害および/または表現型異常を有する患者のルーチンの評価にマイクロアレイ技術を組み込むことにより、多数の反復性の微小欠失/微小重複の迅速な発見がもたらされている。 例えば、わずか2年間の間に、1q21.11,2および16p11.23,4のゲノム領域に対するこれまでに検出されなかった喪失および/または獲得が、自閉症および関連疾患の潜在的な病因因子として広く認識されるようになった。 従来の細胞遺伝学的検査で検出できない染色体領域が小さすぎる臨床的意義のあるコピー数変異体(CNV)が、ヒト核型のすべての染色体対について同定されている。一部のコピー数変異体では、臨床症状の発現および浸透度のばらつきが臨床的意義の確立を複雑にしている。 他の患者では、コピー数変異体とより一貫性のある特異的な表現型所見、および新規の遺伝との関連により、臨床的意義の実証が促進されている。 ここでは、後者の基準を満たす染色体19p13.13のコピー数変異体について述べる。 19番染色体が最も遺伝子に富む染色体の1つ(~2000遺伝子、59 Mb)であるという事実にもかかわらず、19p.5-10を含む欠失を有する個人については、現在までのところ、この微小欠失/微小重複症候群を同定できたのは、4名の臨床遺伝学者による重要な欠失を有する個人の報告の臨床所見と特徴がほぼ一致したことと、それらのマイクロアレイデータが相関していたことであった。
材料および方法
対象
5人の患者がミネソタ大学アムプラッツ小児病院およびミネソタ小児病院の遺伝学クリニックに診断のため紹介された。 Array-CGHは、原因不明の多発性奇形および/または発達障害を有する患者に対する標準的精密検査の構成要素としてオーダーされた。 患者の年齢は最初の検査時に5~26か月の範囲であり、各々4人の臨床遺伝学者の1人によって評価された。 データの共有と結果の公表を可能にするための本試験への参加は、両施設の治験審査委員会により承認された。 これらの患者の両親からインフォームド・コンセントを得た。
アレイベースの比較ゲノムハイブリダイゼーション分析
患者の末梢血由来のDNAを単離し、制限酵素を消化し、ランダムプライマーおよびエキソクレノウ断片DNAポリメラーゼを用いて蛍光色素シアニン5で標識した。 プールされた性別をマッチさせた対照からのDNAを、蛍光色素シアニン3と同時に標識した。 患者および対照のDNAを組み合わせ、アレイベースの比較ゲノムハイブリダイゼーション(aCGH)を、35 Kbの全体の中央値プローブ間隔の平均のバックボーン上の濃縮されたカバレッジの標的領域を含むカスタマイズされた44Kオリゴヌクレオチドアレイを用いて実施した。 最も小さい欠失を有する患者(患者3)の停止点および開始点をより正確に推定するために、平均タイル間隔13 Kbの180Kアレイを実施した(Agilent Technologies, Santa Clara, CA)。 各オリゴヌクレオチドの対照DNAに対する患者の比率は、特徴量抽出ソフトウェア9.1または10.5(Agilent Technologies)を用いて算出し、DNA Analytics 4.0.85(Agilent Technologies)を用いて分析した。 解析に用いた統計アルゴリズムには、ADM1およびADM2が含まれた
(Agilent Technologies) 閾値を対数スケールで0.3の絶対値に設定し、閾値基準を満たす3つの連続したオリゴの要件を備える。
表1 19p13.12-p13.13以内に喪失または獲得した患者の臨床的および細胞遺伝学的特徴
| 患者 | 1 | 2 | 3 | 4 | 5 | Lysyらの患者8 | Auvinらの患者9 | Jensenらの患者10 | Engelsらの患者7 | Strattonらの患者6 |
|---|---|---|---|---|---|---|---|---|---|---|
| 細胞遺伝学的不均衡 | 欠失 19p13.13-p13.2 | 欠失 19p13.13-p13.2 | 欠失 19p13.13 | 欠失 19p13.13-p13.2 | 重複 19p13.12-p13.2 | 欠失 19p13.13-p13.2 | 欠失 19p13.13 | 欠失 19p13.12 | 欠失 19p13.12 | 重複 19p13.13-p13.2 |
| ブレイクポイント(最小) | 12,498,237–13,126,508 | 12,536,641–13,794,080 | 12,793,474–13,104,643 | 12,411,017–13,120,904 | 12,601,112–14,488,238 | 10,256,871–13,188,698 | 12,615,927–13,280,259 | 13,838,264–16,357,778 | 記載なし | 評価なし |
| サイズ(min) | 0.6 Mb | 1.3 Mb | 0.3 Mb (311 Kb) | 0.7 Mb | 1.9 Mb | 2.9 Mb | 0.6 Mb | 2.5 Mb | 2.1 Mb | 記載なし |
| ブレイクポイント(最大) | 12,476,664–13,178,511 | 12,521,732–13,854,243 | 12,779,366–13,120,858 | 12,335,402–13,126,464 | 12,582,355–14,503,887 | 10,246,651–13,280,203 | 記載なし | 記載なし | 記載なし | 評価なし |
| サイズ(max) | 0.7 Mb | 1.3 Mb | 0.3 Mb (341 Kb) | 0.8 Mb | 1.9 Mb | 3.0 Mb | 0.7 Mb | 記載なし | 記載なし | 記載なし |
| 一次試験 | ||||||||||
| 年齢 | 2歳 | 2歳 | 0.75歳 | 0.5歳 | 3歳 | 出生 | 出生時の正常な成長パラメータ | 出生 | 出生 | 出生 |
| 長さ(%ile) | 50th | >95th | 90th | 90th | <5th | <-2 | SD | 記載なし | <3rd | 25–50th |
| 体重(%ile) | 75th | >95th | 50–75th | 90th | 25th | <-2 | SD | <1st | 記載なし | 20th |
| 後頭前野の円周(%ile) | >97th | >95th | >98th | >98th | <-2 SD | -2 | SD | 記載なし | <<3rd | 15th |
| 頭蓋顔面 | 大頭症、前頭隆起 | 大頭症、前頭隆起、下がり気味の口蓋裂 | 大頭症、前頭隆起、下がり気味の口蓋裂 | 大頭症、前頭隆起 | 小頭症、前頭隆起、下がり気味の口蓋裂 | 複雑な頭蓋骨癒合症、前頭の膨らみ、小さな鼻、低い鼻梁 | マクロセフ、高く、大きな額、平らな人中、小さな口 | 扁平な後頭部、高い額、下垂したPF、長い舌、短い鼻 | マイクロ/ブラッキー、薄い上唇、長い人中、小さな口 | マイクロセフ、両側の隆起、上向きのPF |
| 眼科 | 内斜視、眼振、固定不良 | 視神経低形成、外斜視 | 視神経低形成、外斜視 | 視神経萎縮、外斜視 | 視神経低形成、外斜視、眼振 | 眼窩形成不全、両眼隔離症、眼球突出、斜視 | ボタンなし | 斜視、視神経乳頭カッピング、眼瞼下垂、内眼角冗長皮 | 内眼角冗長皮 | 断続的な眼球突出 |
| GI | 慢性下痢 | 腹部の痛み、嘔吐、給餌不良 | なし | 腹部の痛み、嘔吐、セリアック病 | 腹部の痛み、嘔吐、FTT(Gチューブ) | 記載なし | 重度の便秘 | 記載なし | 記載なし | 出生後の疝痛 |
| 脳 | 正常 | 軽度の萎縮、前頭葉 | 吻側脳梁がない、嚢胞性病変 | シリンクスを伴うキアリI奇形 | 正常な磁気共鳴画像 | 中程度の脳室拡大 | 通常の磁気共鳴画像 | 軽度の脳梁および小脳実質の低形成、前期破水 第4通気口 | 通常 | 通常の頭部CT |
| 発作 | – | – | + | + | + | 記載なし | + | 記載なし | 記載なし | 記載なし |
| 筋緊張低下 | + | – | + | + | + | + | 記載なし | + | 記載なし | |
| 発達遅延 | フルスケールIQ49 | 3年間で18か月のスキル。顕著な言葉の遅れ | ほとんど非言語的 | 中程度の言葉の遅れ、特別な教育が必要です。 | 4.5年で21〜29か月のスキル。顕著な言葉の遅れ | 33カ月でした。3.7年で言語なし | 2年で土曜日「精神運動遅延」 | フルスケールIQ63 | 16か月でクロール。スピーチなし | モーターのマイルストーンに到達しなかった、4か月 |
| 最終試験 | ||||||||||
| 年齢 | 6歳 | 3歳 | 14.5歳 | 9.5歳 | 7歳 | 3.7歳 | 2歳 | 記載なし | 1.5歳 | 0.75歳 |
| 伸長(パーセンタイル) | 86th | >95th | 50th | 75th | <3rd | -3 SD | >+3 SD | 記載なし | <3rd | 50th |
| 体重(パーセンタイル) | 55th | >95th | 10–25th | 50–75th | 50th | -0.9 SD | +2 SD | 記載なし | <3rd | 5th |
| 後頭前野周長(パーセンタイル) | >97th | >95th | >98th | >98th | -2 SD | -0.7 SD | +2.5 SD | 記載なし | <<3rd | <5th |
蛍光in situハイブリダイゼーション分析
蛍光in situハイブリダイゼーション(FISH)を実施して、発端者における欠失および/または重複の存在を確認し、もし発見されれば再発リスクに有意な影響を与えるであろう両親における染色体間再配列を除外した。 FISHプローブのために選択されたBACは、RP11-654K9(MAST1を含む)、RP11-963I8(NFIXを含む)、RP11-14L14(CACNA1Aを含む)、および患者5のためにRP11-56K21(PKN1を含む)であった。 RP11-654K9、RP11-14L14、およびRP11-56K21のためのスタブは、CHORI BAC/PACリソースから入手し、クロラムフェニコールを用いてLuriaブロスプレート上で増殖させ、ニックトランスレーション(Abbott Molecular, Abbott Park, IL)により標識した。 Enzo-Greenで予め標識されたRP11-963I8は、Empire Genomics(Empire Genomics, Buffalo, NY)から入手した。 19qサブテロメア領域(D19S238E; Abbott Molecular)に対するプローブも対照として使用した。 欠失の有無を評価するために、10個の中期細胞をスコア化し、重複の有無を評価するために、75~100個の間期細胞をスコア化した。
結果
臨床所見
各患者の詳細な病歴および所見を以下に示し、表1に要約する。
患者1
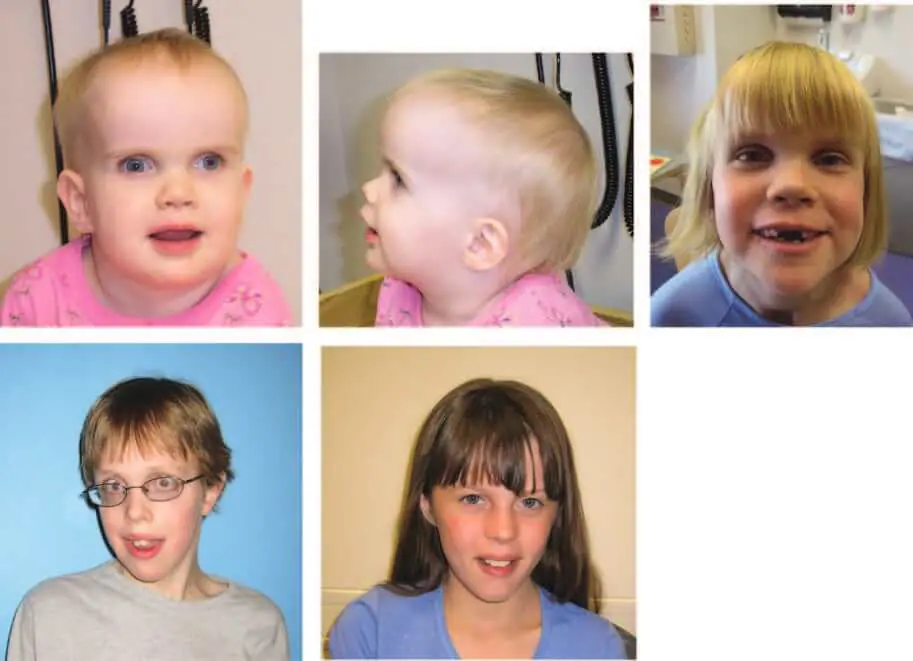
この女児は正期産(出生時体重4026g)で生まれた(図1)。 新生児期の異常には黄疸と舌小帯の切断を要する母乳育児不能があった。 彼女の両親は、生後3か月の時点での彼女の発達に懸念を持っていたが、それは彼女が年をとるにつれて明らかになり、時間の経過とともに肉眼的運動および言語の遅れを含んでいた。 眼科学的および視覚的な問題には、内斜視、ぴくぴく動く眼運動、末端注視眼振、および不十分な固定が含まれた。 患者は視力の成熟が遅れ、経時的に改善し、斜視を矯正するために2回の手術を受けた。 病因不明の慢性下痢を特徴とする消化管障害が最初の4年間存在した。 22か月後の身体診察では、巨頭症(>97パーセンタイル)、50パーセンタイルでの身長、75パーセンタイルでの体重が示された。 鼻橋の陥没、鼻先の上向き、口蓋が比較的高アーチ状である小さな口に加えて、眼窩上隆起および深部セット眼を伴う顕著な前頭隆起が認められた。 彼女の指と足指は長く、足の裏に深いしわがあった。 患者は低緊張性で、深部腱反射は良好であった。 30か月後の検査では、身長と体重は80パーセンタイルを超え、後頭前頭周囲>97パーセンタイルであったが、22か月後にはわずか0.3cm大であった。 6歳時の追跡調査では、86パーセンタイルの身長、55パーセンタイルの体重、97パーセンタイルを超える後頭前野の円周1 SDが示された。 ウェクスラーの最新の評価では、就学前のバッテリーの言語IQは61、パフォーマンスIQは47、フルスケールIQは49であった。 彼女の細かい運動能力と視覚運動能力は繰り返し評価され、比較的遅れている。頭蓋と骨年齢の放射線学的検査は、X線およびCTで正常だった。 脳の磁気共鳴画像(MRI)は視交叉前視神経の軽度低形成を示したが、脳は構造的に正常だ
った。 心エコーおよび腎超音波は正常であった。 臨床検査では、正常な46、XX核型(550バンドレベル分解能)、正常な尿代謝スクリーニング、および正常な血清アミノ酸、アンモニア、乳酸、およびピルビン酸が示された。
患者2
この女児は正期産(出生時体重2835g)で生まれた。 6か月後、肉眼的および微細な運動発達遅延が認められ、視覚的に焦点を合わせ、追跡することができなかった。 患者は46.5cm(>95パーセンタイル)の後頭前野の円周を有する巨頭症であり、下向き眼瞼裂を有していた。 生後7か月の乳児発達のベイリー乳幼児発達検査は3か月で、肉眼的運動レベルは2~5か月、微細運動レベルは1か月であった。 15か月後の脳波は正常で、聴力も正常であった。 17か月で、患者は嘔吐に関連した慢性および一過性の腹痛を呈した。 脳の磁気共鳴画像は軽度の萎縮、特に前頭葉の萎縮を示した。 外斜視が続き、眼科検査で両側視神経低形成が示された。 心音および腎音は正常であった。 細胞遺伝学的分析は正常な46,XX核型を示した。 臨床所見はソトス症候群を示唆するため、NSD1の配列決定を行い、結果は陰性であった。 追跡評価では、言語および発達の遅れが持続し、3歳時の能力レベルは約18か月であり、言語遅延(言語失行)は有意であり、後頭前野の円周が95パーセンタイルを超える持続性大頭症であることが示された。
患者3
この少年は出生時体重3884gで出生し、乳児期に大型の巨頭症を呈し、広い額、前頭部隆起、下斜めの眼瞼裂、筋緊張低下、および顕著な発達遅延を呈した(図1)。 患者は4歳時にてんかんを発症し、医学的にコントロールされている。 小斜角斜視と上眼瞼後退(くぼんだ眼窩と瞬目減少)を矯正するため、2回手術を受けた。 乳児期から14歳まで、彼の巨頭症は持続したが(後頭前野の円周>98パーセンタイル)、身長は90パーセンタイルから50パーセンタイルへ、体重は50~75パーセンタイルから10~25パーセンタイルへと徐々に低下した。 眼科的追跡では、8歳(磁気共鳴画像による正常視神経)から11歳(球後、視交叉前、視交叉視神経低形成)の間に発症した視神経萎縮が認められた。 最近の検査(年齢14.5歳)では、身長は25~50パーセンタイル、体重は10~25パーセンタイル、頭囲は98パーセンタイルであった。 変形の特徴
顔面の緊張が低く、耳が大きい、口蓋が高く、歯が密集している、手が大きい、足が平らであるなどであった。 彼は重度の発話遅延があり、いくつかの選択された言葉を除いて言葉はなかった。 脳の磁気共鳴画像は原因不明の慢性後頭嚢胞性病変と脳梁の吻側の欠如を示した。 連続的な腎超音波検査では、安定したままの腎盂外と解釈される左腎盂の拡張または隆起が認められた。 細胞遺伝学的検査では、正常な46のXY核型(550バンドレベルの解像度)が示され、分子検査ではNSD1またはFMR1の欠失または突然変異は陰性であった。
患者4
この女児は正期産(出生時体重4097g)で、乳児期に低緊張、巨頭症、大きな青色虹彩を伴う目立つ眼球、外斜視を呈し、視神経蒼白が1年以内に認められた(図1)。 その他の臨床的所見としては、上向きの鼻、低い顔面トーン、6つの三日月状のカフェ・オ・レ(cafe-au-lait)斑、ミオクロニー(myoclonic jerks)などがあり、9歳の時に1回の遷延性発作を起こした。脳波ではてんかんは検出されなかった。 2~3年時の歩行、2年前の言葉なしの構音障害を含む運動および言語遅延が認められた。 4年目に斜視手術を受けたが、早期の視覚不注意は経時的に改善した。 7~9年の間にセリアック病の徴候を発症し、グルテンを含まない食事をしていた。 最新の9.5歳時の診察では、彼女は特別な教育サービスを必要とするが、語彙は著しく拡大した。 5か月後の脳の磁気共鳴画像は正常であったが、4年後の反復検査では、キアリI型奇形(脊髄空洞症、5歳時に外科的に減圧)および視交叉および頭蓋内視神経の低形成が示された。 臨床検査では、代謝検査(血清アミノ酸、血中長鎖脂肪酸、尿中有機酸、筋生検、およびミトコンドリアDNA検査)、細胞遺伝学的検査(750バンドレベルの解像度)、およびNF1突然変異分析の結果は正常であった。
患者5
この少年は在胎36週(出生時体重2835g)で生まれた。 2か月後、摂食障害、便秘、頻回の嘔吐、著明な易刺激性、多発性感染症を呈し、同胞より淡い顔色を示した。 14か月後の体重、身長、後頭前野の円周はいずれも5パーセンタイル未満であった。 その他の臨床所見としては、額の傾き、鼻翼の狭さ、乳頭の逆転、殿部の異常な脂肪分布などがあった。 患者はてんかんを有し、左前頭側頭葉の焦点から毎日約4~5回の発作が起こった。 摂食困難と口腔摂取嫌悪が続いたため、Gチューブ留置を伴うNissen噴門形成術を施行した。 眼科検査で水平眼振を認めた。 3年までに、彼の頭囲は5パーセンタイルより有意に低く維持され、持続的な発育不全と発達遅延(わずか数語、2歳時に歩行)を伴った。 4.5歳時の神経心理学的検査では、21~29か月齢の同等の発達がみられ、活動亢進、睡眠障害(1晩に約3時間の睡眠を伴う遅延睡眠開始)、かんしゃく、強迫行動および自傷行動がみられた。 彼の受容的言語は軽度に障害され、表現的コミュニケーションは有意に障害され、最初は6歳時に文章で話した。 過去2~3年で若干の改善が見られる。 患者の成長速度は遅く(3.2cm/年、<1パーセンタイル)、現在、7歳時の身長は3パーセンタイル未満であるが、彼の口腔摂取嫌悪が改善するにつれて体重は50パーセンタイルまで増加した。 家族歴は、患者の父親が眼振を有し、学習困難を報告する点で有意であった。 放射線学的検査には、胸部X線、腹部超音波、上部消化管、軟性S状結腸鏡検査、脳の磁気共鳴画像があり、いずれも正常であった。 電解質、分画を伴う全血球算定、肝臓および甲状腺機能検査、免疫グロブリン、発汗塩化物、血清アミノ酸、および尿中有機酸などの臨床検査はすべて正常と報告された。 細胞遺伝学的分析は、850バンドレベルの分解能で正常な46,XY核型を示した。FISH検査(蛍光 in situ ハイブリダイゼーション法 )は、ウィリアムス症候群、テロメアFISH、および外部の基準検査室で実施されたBAC aCGHは正常であった。
アレイ-CGH分析
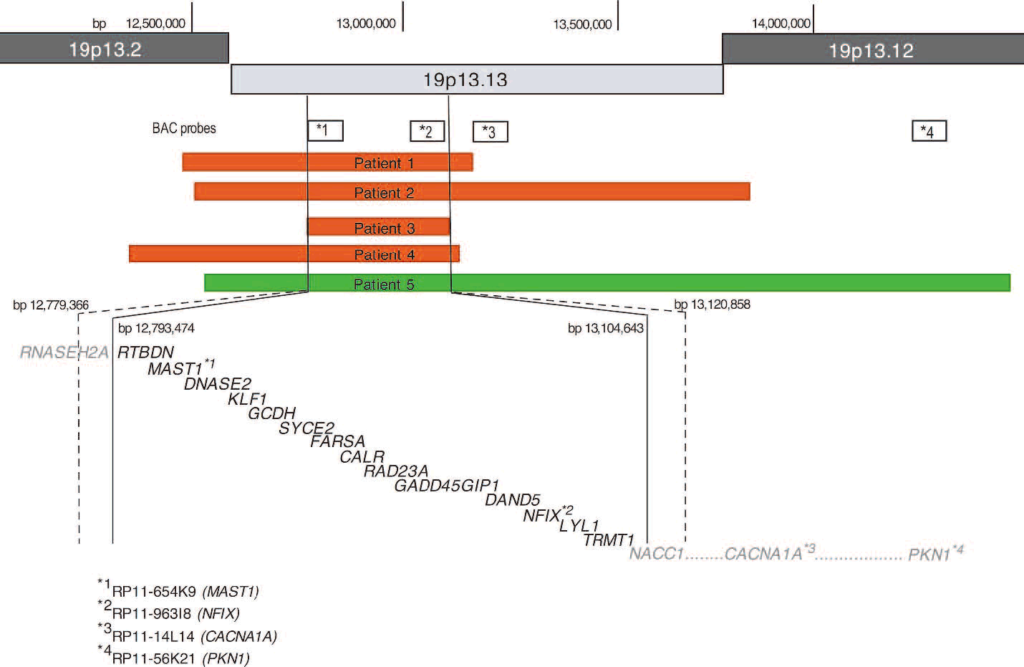
Array-CGHでは、患者1~4の喪失領域と一致する比プロファイルが示され、患者5のゲイン領域が示された。 図2に示すように、喪失領域は約0.31 Mb(患者3)から1.3 Mb(患者2)の範囲であった。 患者5のゲイン領域は~1.9 Mbであった。 患者3にみられる欠失によって区切られたオーバーラップの最小領域(SRO)は~311 Kbに及び、バンド19p13.13に局在する。 Human Genome Build 18に基づくと、このSROの開始点と停止点は、12793474と13104643(最小ブレークポイント)、1279366と13120858(最大ブレークポイント)であった。 全患者の両親にaCGHの追跡調査を行った(図3)。 19p13領域の比プロファイルは、親標本の各々について正常範囲内であった。
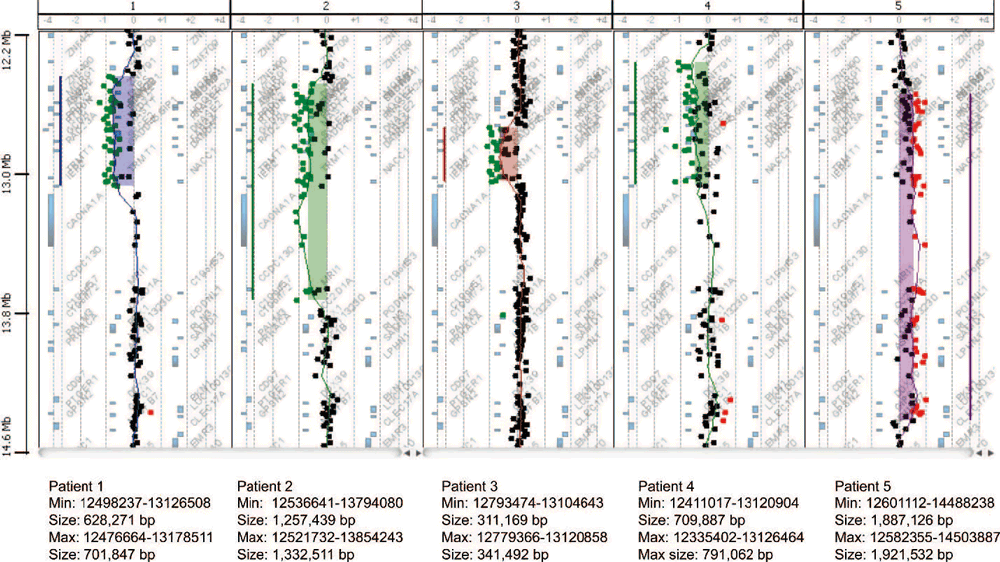
図3. 各患者の両親から得られたアレイ比較ゲノムハイブリダイゼーション(aCGH)プロファイル(中央は参考のための患者アレイ)では、関心領域に不均衡は見られない。

FISH分析
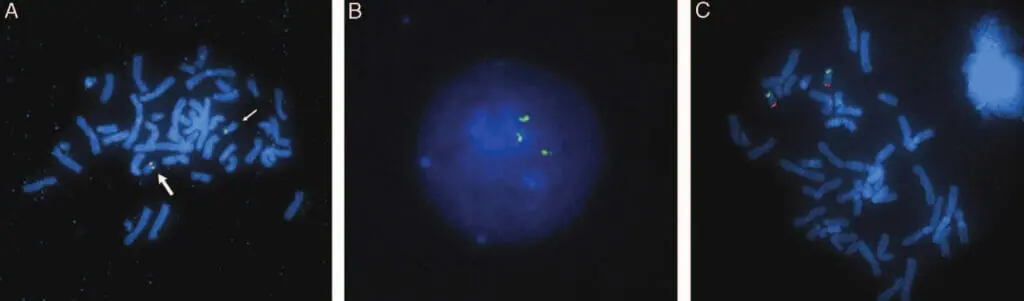
患者1~4におけるコピー数の減少は、間質性欠失を表すものとしてFISHによって確認された。 aCGHの結果と一致して、全例がRP11-654K9(図4A)およびRP11-963I8の両方の欠損を示したが、患者2のみがRP11-14L14の欠損を示した。 コピー数の増加を伴う患者5は、重複と一致する間期シグナルパターンを示した。 プローブRP11-654K9、RP11-963I8、RP11-14L14およびRP11-56K21について、間期細胞のそれぞれ88%、78%、80%および65%は、3つのハイブリダイゼーションシグナルを示した(図4B)。 中期染色体上のFISHでは複製は視覚化できなかったが、シグナルは19番染色体上にのみ存在し、余分なシグナルがゲノムの他の場所に挿入された可能性は否定された。 親FISH試験では、欠失または重複の証拠は示されず、すべてのBACシグナルが#19染色体に局在することがさらに確認され、染色体間挿入の可能性は除外された(図4C)。 したがって、発端者の損失と利益は、新たな出来事を表している。SROに含まれる遺伝子:患者3で発見された消失領域で区切られたSROは 患者3で発見された欠損領域で区切られたSROには RNASEH2A(テロメリック)からNACC1(セントロメリック)までの16個の遺伝子の全部または一部を含み、RNASEH2のほとんど RNASEH2の大部分、RTBDNの全て、MAST1、DNASE2、KLF1、GCDH, syce2, farsa, calr, rad23a, gadd45gip1, dand5, nfix, LYL1とTRMT1、そしてNACC1の大部分である(図5)(http://genome.ucsc.edu; UCSC hg18 Mar 2006)。11,12
図4. A. 患者3のメタフェース細胞にBACクローン(RP11-654K9、赤、RP11-14L14、緑)を組み合わせてFISHを行った。正常な19番染色体(大矢印)は両方のプローブにハイブリダイズし、黄色の融合シグナルが得られた。欠失した19番染色体(小矢印)は、RP11-14L14のみにハイブリダイズし、赤色のシグナルが出なかった。B. 患者5の間期細胞では、RP11-56K21のプローブに3つのシグナルが見られる。C. RP11-963I8(緑)とD19S238E(赤)のプローブを用いて正常なシグナルパターンを示す親検体のメタフェース細胞。染色体間の再配列は見られない。
考察
aCGHにより、19p13.12~19p13.2を含む微小欠失および微小重複を有する4名の患者が同定された。 SROは、19p13.13内に約311 Kb(最大340 Kb)の最小の欠失を伴う患者3によって区切られた。 最小のSROは、約12793474bpから13104643bp(Build 18)に及ぶ。現在までのところ、このSROを包含する欠失を有する患者(Auvinら9により報告された患者)に関する発表された症例報告は2件のみである。 約740 Kbの欠失を伴い、1件はLysyら8により報告された。 より大きな3 Mbの欠失を伴う。 本試験およびAuvinらの試験の患者 この19p13.13の臨床的-細胞ゲノム実体を、微小欠失/微小重複症候群として指定することを支持する一貫した表現型相関を示す。 Lysyらの患者8 しかし、この欠失は本報告のテロメアにほぼ3 Mbのテロメアを及ぼし、最大欠失のほぼ3倍、臨界領域のほぼ10倍の大きさであることから、表現型は有意に異なると考えられる。
19p13.13欠失患者における臨床所見は、3つの反復性異常の一群で注目に値する。 第一に、過成長である。 患者1~4およびAuvinらの患者9 これらの患者はいずれも年齢に応じて大頭症であり、身長および体重は頭蓋顔面の大きさと一致していた。 患者2および3、ならびにAuvinらの患者9にとって、特に興味深いことである。 Sotos症候群を除外するため、NSD1検査を実施した。 したがって、この19p13.13の領域は、Sotos症候群が鑑別診断を受けているがNSD1検査の結果が陰性である他の患者に関連している可能性がある。 2番目によくみられる臨床症状は、眼科的異常(特に斜視)および視神経萎縮または低形成であり、後者は正式な眼科検査および/または磁気共鳴画像によって検出された。 3つ目の所見は、胃腸症状、特に腹痛および嘔吐である。 この三徴にみられる唯一の所見は19p13.13の異常に特異的なものではないが、この一連の所見は、他の微小欠失/微小重複に関する現在数多くの報告の中で報告されているものではない。 しかしながら、これらの19p13.13患者はいくつかの非特異的所見も示した。神経学的異常(例、痙攣、ミオクローヌス性痙攣、筋緊張低下)はいくつかに見られ、発達遅延および/または精神遅滞が全員に存在した。 比較のために、19p13.12-19p13.2内に微小欠失または微小重複を有する他の数少ない発表済み患者を表1に含める(参考文献も参照のこと)。 6、7、10。 これらはいずれも患者3によって区切られた臨界領域と重複せず、それらの表現型にはいくつかの顕著な違いがある。 我々の欠失患者およびAuvinらの欠失患者9とは対照的に これらの重複しない欠失患者7, 10は年齢に応じて小さく、我々が定義した310 KbのSRO内の遺伝子が大きな体の大きさの原因であるという結論を支持している。 我々の欠失患者では難聴はないが、これらの重複しない19p欠失については重大な難聴があり、これらの近位および遠位領域内の遺伝子がそれらの表現型のこの側面を説明することを示唆している。 しかしながら、いくつかの類似点が存在する(例えば、斜視6, 10および視神経乳頭カッピング10)。
重複が認められたのは1例のみであり、臨床症状の一般化には慎重である。 17p11.2および22q11.2を含む、現在よく確立されている相互の微小欠失/微小重複症候群では、各欠失に関連する一連の表現型所見が、対応する重複に関連するものとは十分に異なっており、それぞれ別個の症候群(例えば、17p11.2の欠失についてはSmith-Magenis症候群、17p11.2の重複についてはPotocki-Lupski症候群)としての指定を正当化することが明らかである。
さらに、Wiliams-Beuren遺伝子座が関与する不均衡で認められているように、重複のある患者は欠失のある患者にみられる所見とは逆の所見を有する可能性がある。13, 14 このシリーズでは、大頭症の19p13.13欠失患者と小頭症の重複患者との頭囲の著しい違いによって、このことが同様に示されている。 これは、Strattonら6が報告した重複患者にも当てはまった。 しかし、同じ領域の欠失および重複も、22q11.2の欠失および重複の両方における口蓋咽頭機能不全の所見によって示されるように、いくつかの共通の特徴を共有している可能性がある。 同様に、19pの症例では、欠失や重複の有無にかかわらず、いくつかの臨床所見(眼所見、消化管症状、けいれんなど)が認められた。15-17
SROにマップされた16の遺伝子は、転写、DNA修復、造血などの多様な過程に関与している。 DNASE218とKLF119, 20は胎児の造血に関与する。 1つの機能的KLF1対立遺伝子の存在はヒトの赤血球生成に十分であるため、これら5人の患者に顕性の血液学的異常がないことは予想外ではない。 この領域内の他の遺伝子は、常染色体劣性疾患に関与している: GCDHおよびグルタル酸血症/酸尿症(GA‐1)21-24 およびRNASEH2AおよびAicardi‐Goutieres症候群。 非欠失ホモログの突然変異を伴わない限り、これらの遺伝子の欠失は臨床症状と関連しないと予想される。 SROの遺伝子のいくつかは脳および神経組織で発現し、臨床症状発現の候補遺伝子である。 微小管結合セリン/トレオニンキナーゼ1であるMAST1はそのような候補である。 それは、脳で高度に発現される微小管関連セリン/トレオニンキナーゼファミリーのメンバーである。 特に興味深いのは、MAST1がPTENと相互作用し、PTENの安定化を助けるという事実である。PTENの変異は大頭症と自閉症の両方に関連している。 NF1転写因子ファミリーのメンバーであるNFIXは、別の候補遺伝子である。26-28 マウスモデルは、Nfixが正常な脳の発達に必須であることを示しており、1件の実験では片側接合性が脳梁の無形成をもたらすことが示されており、30 この所見は患者3にみられる所見と類似している。 上述のように、消化管の問題は我々の患者において顕著であった。 これらの知見に興味深い遺伝子はCALRである。CALRは、小胞体内でカルシウムと結合・貯蔵し、核内で働き、核ホルモン受容体による遺伝子転写の調節に作用するタンパク質であるカルレチクリンをコードする。 興味深いことに、それはヒト小腸のニューロンにも局在しており、従って、我々の5人の患者のうち4人に見られる消化器症状に関与している可能性がある。 Auvinら9を含む19p13の欠失に関する以前に発表された報告において、CACNA1AおよびCC2D1Aは、結果として生じる表現型において重要な役割を果たすと仮定されている。9, 31 CACNA1AもCC2D1Aも我々の重要な310~340 Kb領域には含まれていないため、これらは我々の患者におけるこの19p13.13欠失/重複に関連する特異的な表現型コンステレーションの候補とは考えられない。
潜在的候補遺伝子MAST1およびCALRをさらに調査するために、マウスモデルおよび突然変異スクリーニングが提案されている。 前述のように、欠失および重複に関与する開始点および停止点は、各患者に固有であった。 この事実は、欠失/重複領域内に低コピーリピートまたは分節的重複が存在しないこと、または欠失/重複領域に隣接していることとあいまって、19p13 コピー数変異体を生じさせるメカニズムとして非対立遺伝子相同組換えに反対する。 最近では、鎖切断の修復に関与する切断点接合部の微小相同性が、非反復性切断点を伴うコピー数変異体の形成に関与していることが示唆されている。32, 33 しかし、この可能性をさらに検討し、基礎となる分子機構をさらに特徴づけるためには、切断点の詳細な配列決定が必要であろう。応用ゲノム研究センター(TCAG) (http://projects.tcag.ca/variation/)34 のゲノム変異体データベース(DGV) は、細胞遺伝学者や、対照集団で十分に報告されており、良性変異体である可能性のあるコピー数変異体の同定に役立つアレイを扱う他の研究者にとって非常に貴重なものである。 DGVでは、この領域の一部(開始点から12919491まで)がWongら35によって同定されていることに留意されたい。 BACアレイに基づくと、95人中3人で削除された。 特に、文献で発表され、DGV(4000人を超える対照個体を代表する)で引用された大規模対照コホート36-40の中で、この領域の大部分の欠失または重複を同定したものは1つもない。 さらに、我々の症例における欠失および重複がde novoであることが証明されたため、DGVにおける2007年の単一登録は、このコピー数変異体が臨床的に重要であることを裏付けるデータを上回るものではない。 2007年のBACアレイで報告されたコピー数変異体のブレイクポイントを検証することも、さらなる洞察を提供する可能性がある。
要約すると、臨床遺伝学者と細胞遺伝学者の緊密な協力、および遺伝子型と表現型のデータの相関は、19p13.13の微小欠失/微小重複症候群の同定を容易にした。 このことから、同定された患者に対する診断的精密検査(例えば、斜視を検出するための完全な眼科検査および視神経の磁気共鳴画像を含む徹底的な評価、脳の構造的異常を検出するための磁気共鳴画像、後頭前頭周囲の連続的モニタリング、摂食不良、腹痛、嘔吐を含む消化管症状の既往歴の解明)のほか、本症候群のさらなる機能および臨床的特徴との関係を解明する研究の対象となる遺伝子の同定に至った。
謝辞
著者らは、本試験に積極的に参加してくださった患者およびその家族に感謝する。 また、Heather Zierhut, MS, Genetic Counselorの本プロジェクトへの支援に感謝するとともに、ミネソタ大学医療センター、FairviewのCytogenetics Laboratoryの技術者のアレイ-CGHアッセイの実施に感謝する。
参考文献
- Mefford HC, Sharp AJ, Baker C, et al. 染色体1q21.1の再発性再構成および様々な小児表現型。 N Engl J Med 2008;359:1685-1699。
- Brunetti-Pierri N, Berg JS, Scaglia F, et al. 小頭症または大頭症、発達および行動異常に関連する反復性の1q21.1欠失および重複。 Nat Genet 2008;40:1466-1471。
- Weiss LA, Shen Y, Korn JM, et al; Autism Consortium. 16p11.2における微小欠失と微小重複と自閉症との関連 N Engl J Med 2008;358:667-675.
- Marshall CR, Noor A, Vincent JB, et al. 自閉症スペクトラム障害における染色体の構造的変異。 Am J Hum Genet 2008;82:477– 488.
- Hurgoiu V, Suciu S. 19p-の出現。 Ann Genet 1984;27:56-57。
- Stratton RF, DuPont BR, Olsen AS, Fertitta A, Hoyer M, Moore CM. 間質重複19p。 Am J Med Genet 1995;57:562–564.
- Engels H, Brockschmidt A, Hoischen A, et al. DNAマイクロアレイ解析により、原因不明の精神遅滞の候補領域および遺伝子が同定される。 神経学2007;68:743-750。
- Lysy PA, Ravoet M, Wustefeld S, et al. 潜在性19p13.2‐p13.13欠失を伴う症候性頭蓋症候群の新しい症例。 Am J Med Genet A 2009; 149A:2564–2568.
- Auvin S, Holder-Espinasse M, Lamblin M-D. Array-CGHによる19p13.13の新規0.7-Mb欠失の検出(精神遅滞および小児痙攣を伴うてんかんに関連するCACNA1Aを含む)。 てんかん2009;50:2501-2502。
- Jensen DR, Martin DM, Gebarski S, et al. 多発性先天異常の小児における新規染色体19p13.12欠失。 Am J Med Genet A 2009;149A:396–402.
- Kent WJ, Sugnet CW, Furey TS, et al. UCSCのヒトゲノムブラウザ。 ゲノム研究2002;12:996-1006。
- Rhead B, Karolchik D, Kuhn RM, et al. UCSC Genome Browserデータベース: アップデート2010 核酸Res 2010;38(データベース発行):D613-D619。
- Kriek M, White SJ, Szuhai K, et al. 発達遅延および/または先天性奇形を有する患者の間で、重複により隣接する(または埋め込まれていない)領域におけるコピー数の変動;相互および部分的Williams-Beuren重複の検出。 Eur J Hum Genet 2006;14:180 –189.
- Somerville MJ, Mervis CB, Young EJ, et al. Williams-Beuren遺伝子座の重複に関連した重度の表現言語遅延。 N Engl J Med 2005;353:1694 -1701.
- Potocki L, Chen KS, Park SS, et al. 重複の分子機構17p11.2-Smith-Magenis微小欠失の相同組換え相互。 Nat Genet 2000;24:84-87.
- Ou Z, Berg JS, Yonath H, et al. 22q11.2の微小重複はしばしば遺伝し、様々な表現型と関連している。 Genet Med 2008;10:267-277。
- Ensenauer RE, Adeyinka A, Flynn HC, et al. 新興症候群である微小重複22q11.2:13人の患者の臨床的、細胞遺伝学的、および分子解析。 Am J Hum Genet 2003;73:1027–1040.
- Kawane K, Fukuyama H, Kondoh G他 マウス胎児肝における確定赤血球産生のためのDNアーゼIIの必要性 科学2001;292:1546-1549。
- Basu P, Lung TK, Lemsaddek W, et al. EKLFおよびKLF2は、胚βグロビン遺伝子発現および原始赤血球生成において代償的役割を有する。 血液2007;110:3417-3425。
- Hodge D, Coghill E, Keys J, et al. 決定的および原始的な赤血球生成におけるEKLFの全体的な役割。 血液2006;107:3359-3370。
- Kolker S, Garbade SF, Greenberg CR, et al. グルタリルCoAデヒドロゲナーゼ欠損症の小児および成人における自然史、転帰、および治療効果。 小児研究2006;59:840-847。
- Zschocke J, Quak E, Guldberg P, Hoffmann GF. グルタル酸尿症I型.J Med Genet 2000;37:177-181における突然変異分析。
- バスケットC, Merinero B, Christensen E, et al. スペインにおけるグルタリル-CoAデヒドロゲナーゼ欠損症:遺伝学的および生化学的に異なる2群の患者の証拠。 小児研究2000;48:315-322。
- Goodman SI, Stein DE, Schlesinger S, et al. グルタル酸血症(I型)におけるグルタリル‐CoAデヒドロゲナーゼ突然変異:30の新規突然変異のレビューと報告。 Hum Mutat 1998;12:141-144。
- Garland P, Qulaishe S, French P, O’Connor V. セリン/トレオニンキナーゼのMASTファミリーの発現 脳樹脂2008;1195:12-19。
- Borurgeron T. A. シナプストレックから自閉症へ。 Curr Opin Neurobiol 2009;19:231– 234.
- Varga EA, Pastore M, Prior T, Herman GE, McBride KL. 自閉症スペクトラム障害、発達遅延、および巨頭症を伴う臨床小児コホートにおけるPTEN突然変異の有病率。 Genet Med 2009;11:111-117。
- Valiente M, Andres-Pons A, Gomar B, et al. PTENの特異的PDZドメインへの結合は、PTENタンパク質の安定性および微小管結合セリン/トレオニンキナーゼによるリン酸化に寄与する。 J Biol Chem 2005;32:28936-28943。
- Campbell CE, Piper M, Plachez C, et al. 転写因子Nfixは正常な脳の発達に必須である。 BMC Dev Biol 2008;8:52.
- Driller K, Pagenstecher A, Uhl M, et al. 核因子IX欠損症は、脳奇形および重度の骨格欠損を引き起こす。 Mol Cell Biol 2007;27: 3855–3867.
- Basel-Vanagaite L, Attia R, Yahav M, et al. CC2D1Aは、C2ドメインを有する新しい遺伝子ファミリーのメンバーであり、常染色体劣性非症候性精神遅滞に関与している。 J Med Genet 2006;43:203-210.
- Vissers LELM, Bhatt SS, Janssen IM, et al. まれな病原性微小欠失および縦列重複は、微小相同性を介し、局所ゲノム構造により刺激される。 Hum Mol Genet 2009;18:3579 –3593.
- PJ、イラG、ルプスキーJR、ハスティング ヒトコピー数変異の起源に関する微小相同性媒介切断誘発複製モデル。 PLoS Genet 2009;5:e1000327.
- Iafrate AJ, Feuk L, Rivera MN, et al. ヒトゲノムにおける大規模な変異の検出 Nat Genet 2004;36:949-951。
- Wong KK, deLeeuw RJ, Dosanjh NS, et al. ヒトゲノムの共通コピー数変異の包括的な解析。 Am J Hum Genet 2007;80:91–104.
- Redon R, Ishikawa S, Fitch KR, et al. ヒトゲノムにおけるコピー数の全体的な変動。 自然2006;444:444-454。
- Pinto D, Marshall C, Feuk L, Scherer SW. 対照集団コホートにおけるコピー数の変動。 Hum Mol Genet 2007;16:R168–R173.
- Zogopoulos G, Ha KCH, Naqib F, et al. 北米の大集団における生殖細胞系DNAコピー数の変異頻度 Hum Genet 2007;122:345-353。
- Perry GH, Ben-Dor A, Tsalenko A, et al. ヒトのコピー数変異の微細なスケールと複雑なアーキテクチャ。 Am J Hum Genet 2008;82: 685–695.
- Shaikh TH, Gai X, Perin JC, et al. ヒトゲノムにおけるコピー数変異の高解像度マッピングおよび解析:臨床および研究応用のためのデータ資源。 ゲノム研究2009;19:1682-1690。


