 JA
JA
 EN
EN
 中文
中文


記事概要
猫鳴症候群はまれな遺伝性疾患で、乳児期に猫の鳴き声のような高い泣き声を発することが特徴です。この疾患は第5染色体短腕(5p)の一部欠失によって引き起こされ、身体的・発達的・行動的なさまざまな問題を引き起こす可能性があります。早期診断と包括的な介入によって、患者の発達と生活の質を大幅に改善することができます。
この遺伝子座にある疾患に関与する可能性が高い遺伝子

| S/N | 遺伝子名 | 関連する疾患名 | Associated disease name |
| 1 | DLG4 | DLG4 関連シナプソパチー | DLG4-Related Synaptopathy |
| 2 | ACADVL | 極長鎖アシルCoA脱水素酵素欠損症 | Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency |
| 3 | CHRNB1 | 先天性筋無力症候群 | Congenital Myasthenic Syndrome |
| 4 | MPDU1 | N型糖鎖付加と多重経路の先天性疾患 | Congenital Disorders of N-Linked Glycosylation and Multiple Pathways |
| 5 | TP53 | ウィルムス腫瘍素因症候群、リ・フラウメニ症候群 | Wilms Tumor Predisposition; Li-Faumeni Syndrome |
| 6 | WRAP53 | ディスケラトーシス先天性および関連するテロメア生物学的疾患 | Dyskeratosis Congenita and Related Telomere Biology Disorders |
| 7 | GUCY2D | レーバー先天性黒内障、早発性重度網膜ジストロフィー | Leber Congenital Amaurosis; Early-Onset Severe Retinal Dystrophy |
| 8 | ALOX12B | 常染色体劣性先天性魚鱗癬 | Autosomal Recessive Congenital Ichthyosis |
| 9 | ALOXE3 | 常染色体劣性先天性魚鱗癬 | Autosomal Recessive Congenital Ichthyosis |
| 10 | HES7 | 先天性横隔膜ヘルニア、脊肋骨異形成症 | Congenital Diaphragmatic Hernia; Spondylocostal Dysostosis |
| 11 | TMEM107 | メッケル症候群13・ジュベール症候群 | Meckel syndrome 13・Joubert Syndrome |
| 12 | CTC1 | ディスケラトーシス先天性および関連するテロメア生物学的疾患 | Dyskeratosis Congenita and Related Telomere Biology Disorders |
| 13 | RANGRF | ブルガダ症候群 | Brugada Syndrome |
| 14 | RPL26 | ダイアモンド・ブラックファン貧血 | Diamond-Blackfan Anemia |
| 15 | NTN1 | 先天性鏡動作 | Congenital Mirror Movements |
[1_DLG] DLG4 関連シナプソパチー(DLG4-Related Synaptopathy)

DLG4関連シナプソパチーは、DLG4遺伝子の変異によって引き起こされるまれな遺伝性疾患です。この遺伝子は、脳内で興奮性シナプス機能を調節する重要なタンパク質であるPSD-95(シナプス後密度タンパク質95)を作る働きを持っています。この疾患の特徴には、発達の遅れ、知的障害(軽度から中等度が多い)、および自閉スペクトラム症が含まれます。また、約半数の患者にてんかんが見られ、約40%の患者で運動機能や言語能力の退行が報告されています。
その他の神経学的な症状として、筋緊張の低下(低緊張)、反復動作や運動失調(アタキシア)、ジストニア、震え(振戦)といった運動障害、片頭痛が含まれます。睡眠障害も一般的で、入眠や睡眠の維持が困難なケースがよく見られます。また、斜視、遠視、眼振、大脳皮質盲といった眼の異常が報告されています。てんかん発作、乗り物酔い、疲労により嘔吐が引き起こされる場合もあります。身体的には、関節の柔軟性(37%)や脊椎側弯(20%)が比較的多く見られます。

現在、DLG4関連シナプソパチーを根本的に治療する方法はありません。治療は症状の管理と生活の質の向上を目的としています。患者ごとのニーズに合わせて発達の遅れや知的障害、てんかん、片頭痛、運動失調、ジストニア、視覚障害などに対する治療が行われます。不眠症に対しては行動療法が有効な場合があり、重度の睡眠障害には薬物療法が考慮されることもあります。
この疾患を管理するためには、定期的な診察とモニタリングが重要です。診察では、発作や運動機能、筋緊張、片頭痛などの新しい症状がないかを評価します。また、発達の進行、移動能力、日常生活スキルの確認も行われます。特に子どもが成長する過程で、不安、注意欠如・多動症(ADHD)、攻撃性、自傷行為といった行動面の問題が現れることがあるため、適切に対応することが求められます。眼科検査は年に1回または必要に応じて実施し、発達の退行や異常な脳波がある場合には24時間の脳波検査(EEG)も検討されます。
DLG4関連シナプソパチーは、主にDLG4遺伝子の新生(de novo)病的変異による常染色体優性疾患です。まれに、モザイク型またはヘテロ接合型の親から遺伝することがありますが、家族内での再発リスクは一般的に低いと考えられます。遺伝子検査によって病的変異を確認することで、将来の妊娠に向けた出生前診断や着床前遺伝子診断が可能となります。
DLG4関連シナプソパチーの予後(寿命)は不明ですが、47歳まで生存した報告例があり、成人期まで生存する可能性が示されています。過去には、マルファン様体型や特徴的な顔貌がこの疾患の特徴とされることもありましたが、最近の研究ではこれらが一般的な特徴ではないことが確認されています。この疾患の多くの症例は、DLG4遺伝子変異によるタンパク質機能の喪失に起因しています。症状や重症度は個人によって大きく異なり、主に脳の発達と機能に影響を及ぼします。
【もっと知りたい方へ】DLG4 SHINE Foundation
[2_ACADVL] 極長鎖アシルCoA脱水素酵素欠損症(Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency)

極長鎖アシルCoA脱水素酵素欠損症(VLCAD欠損症)は、ミトコンドリアでの長鎖脂肪酸の分解が正常に行われないまれな遺伝性疾患です。この分解プロセスは、空腹時やエネルギー需要が高まる状況でエネルギーを作り出すために不可欠です。この疾患は、脂肪酸のβ酸化の初期段階を担う酵素をコードするACADVL遺伝子の変異によって引き起こされます。その結果、有害な脂肪酸代謝物が体内に蓄積し、心臓、肝臓、骨格筋などの臓器に損傷を与えることがあります。
VLCAD欠損症は、重症度や発症年齢に応じて3つの主要な型に分類されます。
最も重症な早期発症型(心臓および多臓器不全型)は、生後数か月以内に発症することが一般的で、肥大型または拡張型心筋症、心嚢液貯留、不整脈、筋緊張低下(低緊張)、肝腫大(肝臓の腫れ)、およびケトン体を伴わない低血糖(低ケトン性低血糖)などの症状が見られます。
肝型(低ケトン性低血糖型)は幼児期に発症し、低血糖と肝腫大が特徴ですが、心筋症は伴いません。
後発型(筋型)は思春期や成人期に発症し、運動による筋肉痛、けいれん、運動耐性の低下、および運動や空腹によって引き起こされる横紋筋融解症(筋肉の破壊)などの症状を示します。この型では、症状発現時に低血糖が見られることは稀です。
新生児スクリーニング(NBS)によって、VLCAD欠損症の早期診断が大幅に向上しました。診断時に無症状のケースも多く、早期に管理を開始することで重篤な合併症を予防し、予後を改善することが可能です。VLCAD欠損症の発症頻度は、出生30,000人に1人から100,000人に1人と推定されています。
治療の基本は、代謝危機の予防と症状の管理です。日常のケアには、長鎖脂肪を少なくし中鎖脂肪酸(MCT)を多く含む特別な食事療法が含まれます。これにより、欠損した酵素を迂回し、代替のエネルギー源を提供します。また、空腹による症状を防ぐため、特に乳幼児では頻繁な食事や複雑な炭水化物を含む夜間のおやつが推奨されます。カルニチン、オメガ脂肪酸、ビタミンA、D、Eなどのサプリメントを使用することもあります。過度な運動は筋肉症状を引き起こす可能性があるため、適切な運動管理が重要です。

緊急時には、点滴によるブドウ糖と水分補給で血糖値を安定させ、横紋筋融解症を防ぎます。横紋筋融解症が発生した場合は、十分な水分補給と尿のアルカリ化が腎機能を保護するために必要です。手術が必要な場合には、術前管理を慎重に計画することが求められます。
VLCAD欠損症の効果的な管理には、定期的な監視が不可欠です。成長や発達の進捗、栄養状態の評価が定期的に行われます。また、血液検査で代謝マーカーを監視し、心臓の機能を評価するための心エコー検査や筋肉の健康状態の評価も含まれます。
この疾患は、常染色体劣性遺伝の形式で遺伝します。つまり、両親がそれぞれACADVL遺伝子の変異を持つ場合、子どもが発症する確率は25%です。遺伝子検査によりキャリア(保因者)を特定し、将来の妊娠に向けた出生前または着床前遺伝子診断が可能です。
適切な診断と治療が行われれば、重症型の乳児でも良好な予後が期待できます。スクリーニングや食事療法、医療の進歩により、多くの患者が健康的な生活を送ることが可能となりました。ただし、終生にわたる監視と個別の治療計画が、この疾患を効果的に管理するためには重要です。
【もっと知りたい方へ】SRGNとGMDIによるVLCAD栄養管理ツールキット
[3_CHRNB1] 先天性筋無力症候群(Congenital Myasthenic Syndrome)

先天性筋無力症候群(CMS)は、神経と筋肉の間の信号伝達に影響を与えるまれな遺伝性疾患で、筋力の低下を引き起こします。この筋力低下は、活動とともに悪化する傾向があります。症状は通常、出生直後または幼少期(主に2歳まで)に始まりますが、稀に成人期に発症する場合もあります。CMSは主に骨格筋の筋力低下を引き起こしますが、心筋や平滑筋には影響を及ぼしません。認知能力、協調性、感覚、腱反射は通常正常です。ただし、一部のCMSのサブタイプでは、筋力低下に加えてより複雑な症状が見られることがあります。
CMSの重症度や進行はサブタイプによって大きく異なります。軽度の運動不耐性だけを経験する人もいれば、筋力低下が顕著であったり、発熱や感染症、ストレスによって突然呼吸不全を引き起こす人もいます。新生児では、断続的な無呼吸やチアノーゼなどの呼吸問題、授乳困難、吸う力の弱さ、泣き声の弱さ、全身的な筋力低下が一般的な症状です。一部のケースでは、胎児期の運動不足に起因する関節拘縮(先天性関節拘縮症として知られる)や、細長い顔、狭い顎、高い口蓋などの特徴的な顔立ちが見られることもあります。幼児期以降では、運動発達の遅れ、眼瞼下垂(まぶたが下がる状態)、眼筋の筋力低下、顔面や咽頭の筋力低下、嚥下や発声の困難などが一般的です。
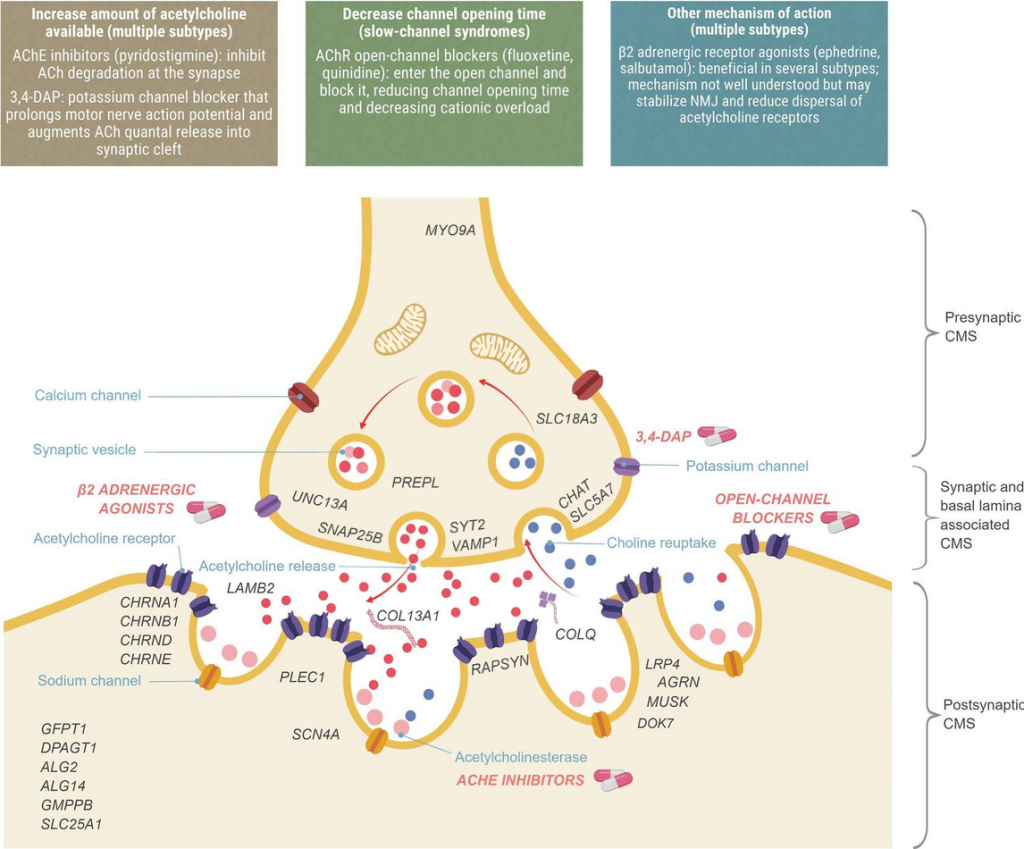
Thompson, R., Bonne, G., Missier, P., & Lochmüller, H. (2019). [Figure 1. Localization of CMS types and therapeutic strategies.] Targeted therapies for congenital myasthenic syndromes: systematic review and steps towards a treatabolome. Emerging topics in life sciences, 3(1), 19–37. https://doi.org/10.1042/ETLS20180100
一部の患者では、リムガードル型筋無力症と呼ばれる特定の筋力低下パターンが見られます。これは、腰部や肩周りの筋肉に影響を及ぼし、よろめくような歩行や階段の上り下りの困難を引き起こします。これらの症状は、ストレス、感染症、発熱などのエピソード中に突然悪化することがあり、慎重な管理が必要です。
CMSの治療は、個々の患者やサブタイプに応じて高度に個別化されます。遺伝子検査は治療方針を決定する上で重要な役割を果たし、異なるサブタイプが異なる治療法に反応します。一般的に使用される薬には、神経と筋肉の間の信号伝達を改善するピリドスチグミン(アセチルコリンエステラーゼ阻害薬)が含まれます。ただし、すべてのサブタイプがこの薬に効果があるわけではなく、一部では症状を悪化させることがあります。他の治療法として、神経伝達物質の放出を増加させる3,4-ジアミノピリジン(3,4-DAP)や、エフェドリンやサルブタモール(特定のサブタイプで有効)が使用される場合があります。薬の副作用は慎重に監視する必要があり、個々の反応に応じて投与量を調整する必要があります。CHRNB1関連CMSの場合、フルオキセチン、キニジン、ピリドスチグミンが第一選択薬として推奨されており、補助的な治療法としてサルブタモールやエフェドリンが挙げられます。薬物治療を始める際は必ず医療専門家に相談してください。
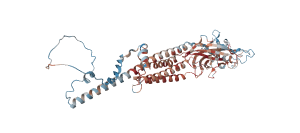
薬物治療以外にも、CMSの管理には生活の質を向上させるための多面的なアプローチが重要です。理学療法や作業療法、言語療法に加え、重症の場合には車椅子や装具、人工呼吸器の使用が検討されることがあります。摂食困難がある患者には、胃ろうが必要になる場合もあります。乳児を持つ親は、無呼吸モニターの使用や心肺蘇生法(CPR)の訓練を受け、突然の呼吸困難に対応できるようにすることが推奨されます。
筋力や呼吸機能、全体的な健康状態の定期的なモニタリングは非常に重要です。子どもは通常6か月ごと、大人は年に1回の評価を受けることが推奨されます。妊娠中の患者は、呼吸および心臓の綿密なモニタリングを行う専門医との連携が必要です。
CMSは免疫応答が原因ではないため、免疫調節療法は効果がありません。ただし、ほとんどのCMSサブタイプは何らかの薬物療法に反応しますが、治療の有効性はサブタイプによって異なります。特定の薬があるサブタイプに有効であっても、別のサブタイプでは悪影響を及ぼす場合があるため、正確な診断と個別化された治療計画が不可欠です。適切なケアと定期的なモニタリングにより、CMSの患者は症状を効果的に管理し、生活の質を向上させることが可能です。
[4_MPDU1] N型糖鎖付加と多重経路の先天性疾患(Congenital Disorders of N-Linked Glycosylation and Multiple Pathways)

N型糖鎖型先天性糖鎖付加異常症および複数経路異常症(Congenital Disorders of N-Linked Glycosylation and Multiple Pathways)は、先天性糖鎖付加異常症(Congenital Disorders of Glycosylation, CDG)として知られています。これらは、糖鎖修飾と呼ばれる、糖分子がタンパク質や脂質に付加される過程に異常が生じることで引き起こされるまれな遺伝性疾患のグループです。この糖鎖修飾は細胞機能にとって非常に重要であり、その異常は多くの臓器に影響を与える多様な症状を引き起こします。
CDGは一般的に乳児期に発症し、典型的な症状として発育不良、発達遅延、筋力低下(低緊張)、肝臓の異常(肝障害)、神経学的な問題(例えばけいれん)が挙げられます。また、眼の異常、皮膚の状態(例えば魚鱗癬)、免疫系の機能不全も一般的です。サブタイプの1つであるMPDU1-CDG(別称:CDG-If)は、糖鎖修飾の重要な過程である糖分子の細胞膜を越えた輸送に関わるMPDU1遺伝子の異常によって引き起こされます。
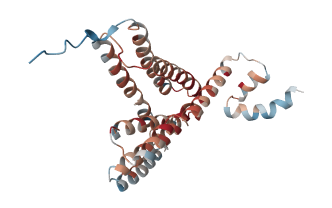
MPDU1-CDG(CDG-Ifとも呼ばれる)は、糖鎖付加障害(CDG)の中でも非常にまれなタイプの遺伝性疾患です。CDGは糖タンパク質の合成過程に異常が生じることで引き起こされる多系統性の遺伝性疾患群です。この異常により血清中の糖タンパク質が十分に糖鎖化されず、発生過程、細胞分化、細胞機能の維持に重要なN型糖タンパク質の働きが損なわれるため、全身にわたる影響が現れます。その結果、多彩な症状が現れるのが特徴です。
MPDU1-CDGは、MPDU1遺伝子における変異が原因で発症します。この遺伝子は細胞内で特定の糖分子を輸送する役割を持っており、糖鎖化の重要なステップを担っています。この機能異常により、適切に糖鎖化されたタンパク質や脂質が生成されなくなり、発達の遅れ、筋緊張低下(低筋緊張)、けいれん、乾燥や赤みを伴う皮膚(魚鱗癬など)、および顔の特徴的な変形といった症状が現れることがあります。また、一部の患者では無呼吸がみられるため、呼吸管理が重要です。
MPDU1-CDGの重症度や進行の仕方は個々の患者や遺伝子変異の種類によって大きく異なります。一般的には、精神運動発達の遅れや神経学的な障害がみられるほか、血液凝固異常、免疫不全、皮膚の異常などの全身的な問題が現れることがあります。このように、糖鎖化異常が神経系、骨格、免疫系を含む多くの系統に複雑な影響を及ぼすことがわかっています。

治療は主に患者の個々の症状に応じた対症療法が中心となります。特に乳幼児期における栄養管理は重要で、摂食困難や成長障害がみられる場合には特殊なミルクや栄養補助食品、経管栄養が必要になることがあります。口腔運動機能の障害による嘔吐が続く場合には、濃いめの食品や制酸剤を使用することで対処することができます。また、言語療法、理学療法、作業療法を取り入れることで、発達遅延や運動機能の改善を図ることが可能です。
目の異常がよくみられるため、眼科医による早期の診察と対応が視力の維持に役立ちます。腎機能や甲状腺機能も合併症として影響を受ける可能性があるため、腎超音波検査や尿検査を定期的に行い、ネフローゼ症候群や蛋白尿などの問題を早期に発見することが推奨されます。また、血液凝固異常が手術時に問題を引き起こすことがあるため、事前の血液学的な相談が必要です。一部のケースでは、血液製剤や血漿輸注が行われることもあります。
急性期の症状、例えば脳卒中様の発作や重度の感染症の場合、静脈点滴や血糖値の安定化などの迅速な対応が必要です。特に無呼吸の管理は重要で、乳幼児の保護者には呼吸器の緊急事態に備えた応急処置の訓練が推奨されます。

MPDU1-CDGを診断するためには遺伝子検査が必須であり、他のCDGサブタイプとの鑑別にも役立ちます。早期診断によって適切な介入が可能になり、予後や生活の質の改善につながります。この疾患はまれであるため長期的なデータは限られていますが、記録された症例では包括的なケアと多職種による管理によって思春期まで生存する患者もいます。
MPDU1-CDGの治療には、神経科、消化器科、眼科、血液科、免疫科などの専門医による連携が不可欠です。この多職種アプローチにより、疾患が引き起こす多岐にわたる課題に対応し、患者の全体的な治療成績の向上を目指します。
[5_TP53] ウィルムス腫瘍素因症候群、リ・フラウメニ症候群(Wilms Tumor Predisposition; Li-Faumeni Syndrome)

リ・フラウメニ症候群(LFS)は、若年でがんを発症するリスクが非常に高くなる稀な遺伝性疾患です。この疾患は常染色体優性遺伝の形式をとるため、罹患した人は自分の子どもに50%の確率でこの疾患を遺伝させる可能性があります。LFSは主にTP53遺伝子の生殖細胞系列変異と関連しており、この遺伝子は細胞分裂を調整し、がんの発生を抑える重要な役割を果たしています。
LFSの特徴として、家族内でがんの発症が多いことが挙げられます。古典的な診断基準では、45歳以前に肉腫(サルコーマ)を発症した患者(プロバンド)がいること、45歳以前に何らかのがんを発症した近親者(1親等)がいること、さらに45歳以前にがんを発症したか、または年齢に関係なく肉腫を発症した別の近親者がいることが条件です。これに類似するが、基準を完全には満たさない場合は「リ・フラウメニ様症候群(LFL)」と呼ばれることがあります。LFSに関連するがんとしては、乳がん、軟部組織および骨の肉腫、脳腫瘍(アストロサイトーマなど)、副腎皮質がんが代表的で、TP53変異を持つ人のがんの約80%を占めます。他にも白血病、大腸がん、胃がん、ウィルムス腫瘍、悪性葉状腫瘍、脈絡叢乳頭腫などが関連しています。
ウィルムス腫瘍は主に小児に発生する腎臓のがんで、LFSに関連するがんの一つです。通常は健康そうに見える子どもの腹部腫瘤として現れ、時に腹痛、発熱、貧血、血尿、高血圧といった症状を伴うことがあります。ウィルムス腫瘍の5〜10%は両側性または多発性で、LFSのような遺伝的素因を持つ場合、その発生率が高くなる傾向があります。診断には組織学的解析が必要で、腫瘍は良好型と退形成型の2つに分類されます。退形成型は通常、体細胞のTP53変異と関連しており、予後が悪い傾向にあります。治療には手術、化学療法、場合によっては放射線療法が含まれます。重篤な疾患であるものの、全体的な生存率は90%以上と非常に高いですが、退形成型や遠隔転移を伴う場合には予後が悪化することがあります。

LFSにおけるがんのリスクは年齢に応じて変化します。小児期には副腎皮質がん、横紋筋肉腫、脳腫瘍(脈絡叢乳頭腫など)が一般的です。青年期から若年成人期には乳がん、肉腫、脳腫瘍が多く、成人後期には膵臓がんや前立腺がんのリスクが高まります。一生涯におけるがん発症リスクは、女性で約90%、男性で約70%とされています。がんの約半数は40歳以前に発症します。また、治療の影響や遺伝的素因により、二次がんのリスクも高くなります。
LFSの管理には、がん予防、監視、治療が含まれます。女性においては、乳がんのリスクを下げるために予防的乳房切除が選択肢となることがあります。監視としては、頻繁な身体検査、全身MRI、個別の家族歴や年齢に応じた臓器特異的検査が推奨されます。放射線療法は二次がんのリスクを高めるため、可能な限り避けられます。遺伝カウンセリングは、罹患家族にとって重要な支援であり、スクリーニングや予防措置の指導を行います。TP53変異の検査により、保因者を特定し、早期介入が可能になります。

妊娠中の女性においては、がんの兆候を早期に発見するための慎重な監視が必要です。TP53変異を持つ胎児の監視に関する特定の推奨事項はありませんが、出生後にスクリーニングを開始することが推奨されます。稀な合併症として、妊娠性絨毛がんが報告されています。
LFSは、個別化医療と積極的な管理の重要性を示しており、罹患した個人や家族の予後を改善するための適切な取り組みが求められる疾患です。
[6_WRAP53] ディスケラトーシス先天性および関連するテロメア生物学的疾患(Dyskeratosis Congenita and Related Telomere Biology Disorders)

ディスケラトーシス・コンジェニタ(Dyskeratosis Congenita, DC)および関連するテロメア生物学障害(Telomere Biology Disorders, TBD)は、テロメアの維持機能に異常が生じることで発症する、非常に稀な遺伝性疾患群です。テロメアは染色体の末端を保護する構造で、遺伝子の安定性と細胞の寿命を保つために重要です。いくつかの遺伝子、特にWRAP53遺伝子の変異がこのプロセスを妨げ、多様な臨床症状や合併症を引き起こします。
WRAP53遺伝子は染色体17p13に位置し、テロメラーゼ・カハール体タンパク質1(Telomerase Cajal Body Protein 1)をコードしています。このタンパク質は、テロメア合成、DNA修復、腫瘍抑制タンパク質p53の調節において重要な役割を果たします。p53経路は、細胞周期の調節やDNA損傷への応答に欠かせない仕組みです。WRAP53遺伝子の変異により、テロメア維持機能が障害される常染色体劣性型のディスケラトーシス・コンジェニタ(DKCB3)が発症します。この疾患は、テロメア維持の異常がどのようにして多くの臓器系に影響を及ぼすかを示す代表例といえます。
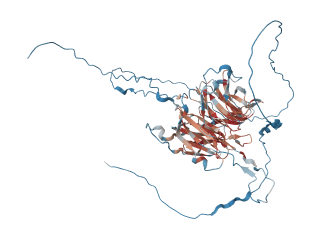
DC/TBDの臨床症状は非常に幅広いです。ディスケラトーシス・コンジェニタ(DC)の古典的な特徴は、胸部や首に見られる網目状の色素沈着、爪の形成不全、口腔内白板症の3つの症状(三徴候)です。ただし、すべての患者にこの三徴候が揃うわけではなく、症状の現れ方には大きな個人差があります。進行性の骨髄機能不全、骨髄異形成症候群、急性白血病、扁平上皮癌(特に頭頸部や肛門生殖器に発生するもの)、肺線維症、肝疾患といった症状が追加的に見られることがあります。他にも、涙目、眼瞼異常(内反や外反など)、タウロドンティズム(歯根の形態異常)、胃腸の毛細血管拡張症、大腿骨や肩の無血管性壊死などが報告されています。髪の早期白髪化、骨粗鬆症、再生不良性貧血なども頻繁に見られますが、その程度は人によって異なります。多くの場合、症状は幼少期(5歳から10歳頃)に発症し、骨髄機能不全、感染症、肺合併症、あるいは悪性腫瘍によって早期に死亡することが多いです。
DKCB3はDCの一形態で、常染色体劣性遺伝のパターンを持ちます。DC全体としては、X連鎖性劣性、常染色体優性、常染色体劣性の3つの遺伝形式があります。この疾患は主に男性に発症しますが、様々な民族グループで報告されています。DC/TBDの根本的な原因はテロメア維持の異常にあり、これが上皮細胞や造血細胞の増殖を妨げ、最終的に細胞老化を引き起こします。DCの特徴である色素異常は、老化したメラノサイトでのメラニン合成の増加が原因とされています。
多系統に影響を及ぼす疾患ではありますが、大部分の患者は正常な運動発達や神経機能を維持しています。ただし、一部のケースでは発達遅延が見られることもあります。

DC/TBDの治療は複雑で、個々の患者に合わせた対応が必要です。骨髄移植(HCT)は骨髄機能不全や白血病に対する唯一の根治治療ですが、長期的な治療毒性を伴うリスクが大きいです。適切なドナーが見つからない場合には、アンドロゲン療法が骨髄機能不全の代替治療として考慮されることがあります。悪性腫瘍や肺線維症など、他の合併症の治療は症状に応じた対応が求められます。肺線維症が重篤な場合には、肺移植が必要になることもあります。ただし、がん治療には長期的な血球減少や臓器毒性といった追加的なリスクが伴います。
DC/TBDの管理には定期的な経過観察が不可欠です。年1回の血球数検査や骨髄検査、がん検診、肺機能検査、肝機能の評価を行い、特にアンドロゲン療法を受けている場合には肝機能や内分泌系のモニタリングが重要です。また、口腔内白板症や二次感染のリスクを減らすために、6カ月ごとの歯科検診と適切な口腔ケアが推奨されます。さらに、喫煙や過剰な日光曝露、一部の薬剤の使用を避けることで、腫瘍発生や合併症のリスクを下げることができます。
DC/TBDは非常に稀な疾患であり、その発生頻度は一般集団では不明です。2022年3月時点で、世界的に記録された症例数は約800~1,000件とされています。このような稀少性から、専門的なケアとさらなる研究が必要とされています。
[7_GUCY2D] レーバー先天性黒内障、早発性重度網膜ジストロフィー(Leber Congenital Amaurosis; Early-Onset Severe Retinal Dystrophy)

レーバー先天性黒内障1型(LCA1)は、染色体17p13.1上に位置するGUCY2D遺伝子の変異によって引き起こされる重度の遺伝性網膜疾患です。この遺伝子は、視細胞(光を感知する網膜の細胞)の棒体および錐体内で重要な役割を果たす「網膜グアニル酸シクラーゼ1(RETGC-1)」というタンパク質を生成します。このタンパク質は、cGMP(環状グアノシン一リン酸)と呼ばれる分子を生成する働きを持ち、網膜で光を電気信号に変換する「視覚伝達(光受容の回復過程)」の重要な役割を担っています。また、視細胞の外節膜へのタンパク質輸送にも関与している可能性があります。RETGC-1が正常に機能しなくなると、視細胞が正しく働かなくなり、網膜が深刻な変性を起こします。
LCA1は通常、生後または幼少期に発症します。この疾患は「レーバー先天性黒内障(LCA)」や「早期発症型重症網膜ジストロフィー(EOSRD)」と呼ばれる病態スペクトラムの一部とされています。LCAの中でも特に重症なLCA1は、生まれつき著しい視力低下や失明を特徴とし、随意ではない眼球運動(眼振)、光に対する瞳孔反応の鈍さまたは欠如、目をこする、押す、または突くなどの行動(眼デジタルサイン)が見られることがあります。また、光過敏症(まぶしさ)、強い遠視、円錐角膜(角膜が円錐状に変形する症状)、そして時間の経過とともに網膜の色素変化などの症状も現れる場合があります。視覚機能を測定する電気網膜図(ERG)では、ほとんどまたは全く反応が検出されないことが一般的です。
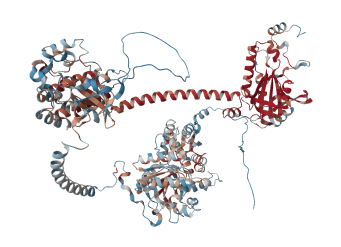
乳幼児期には網膜の見た目が正常に見える場合がありますが、加齢とともに色素性網膜症、血管の狭窄、黄斑萎縮、視神経の蒼白化などの進行性の異常が見られることが多いです。また、視神経乳頭ドルーゼンや水晶体混濁などの合併症が起こることもあります。ただし、LCA1では視覚機能に大きな障害があっても、光干渉断層計(OCT)で確認される網膜構造は比較的よく保存されていることがあります。このため、RETGC-1の機能を回復させる治療が視力改善に有効である可能性が示唆されています。
LCAの発生率は約80,000人に1人とされ、GUCY2D遺伝子の変異は全症例の6〜21%を占めると推定されています。この疾患は常染色体劣性遺伝形式で遺伝するため、両親から受け継いだ2つの遺伝子に変異がある場合に発症します。LCA1の視力は通常、光を全く感じない状態から最大でも0.05(20/400)程度であり、自然な視力回復の可能性は低いとされています。

LCA1および関連疾患の管理には、さまざまな専門分野を統合したケアが必要です。発症早期から発達支援や弱視リハビリサービスを活用することで、より良い結果を得ることが期待されます。教育専門家や理学療法士、作業療法士の支援を受けることや、視覚補助機器を利用することが推奨されます。また、必要に応じて行動療法や発達支援プログラムを組み合わせることも有効です。成長に伴い、就労支援や経済的援助などの追加的な支援を受けることができる場合があります。
近年では、遺伝子治療の分野で有望な進展が見られます。例えば、GUCY2D遺伝子の正常なコピーを視細胞に導入することでRETGC-1の機能を回復させる治療が研究されています。ATSN-101のような新しい治療法は、これまで治療が困難とされてきたLCA1の患者にとって大きな希望をもたらしています。これらの治療によって、視力を保持または改善する可能性が期待されています。
[8_ALOX12B,9_ALOXE3] 常染色体劣性先天性魚鱗癬(Autosomal Recessive Congenital Ichthyosis)


常染色体劣性遺伝性先天性魚鱗癬(ARCI)は、皮膚のバリア形成に関与する遺伝子、特にALOX12BやALOXE3の変異によって引き起こされる稀な遺伝性皮膚疾患の一群です。これらの変異は、皮膚の保護バリアを形成し維持するために必要な酵素の働きを妨げます。ARCIは、生まれつきまたは乳幼児期早期に見られる異常な皮膚の鱗状の剥がれ(角化亢進)や皮膚の赤み(紅斑)が特徴です。この疾患には、重症度や症状の見た目が異なるいくつかの亜型が含まれ、その中にはラメラ型魚鱗癬(LI)や先天性魚鱗癬様紅皮症(CIE)があります。
ALOX12B遺伝子はアラキドン酸12リポキシゲナーゼという酵素をコードしています。この酵素は、皮膚細胞(角質細胞)の脂質エンベロープを形成する上で重要な役割を果たし、オメガ-ヒドロキシセラミドを修飾します。この修飾は、皮膚バリアの水分保持機能や外的刺激からの保護に不可欠です。ALOX12Bの変異によりこれらの過程が妨げられると、ARCI2と呼ばれるARCIの一型が発症します。この型では、生まれたときに薄く光沢のある「コロジオン膜」に包まれていることが多く、この膜は時間とともに剥がれ落ち、LIやCIEに特徴的な皮膚の鱗状化や赤みが現れます。ALOX12Bの変異を持つ多くの人は、細かい鱗状の剥がれと紅斑を特徴とする軽度から中等度のCIEを発症します。
同様に、ALOXE3遺伝子はヒドロペルオキシドイソメラーゼという酵素をコードしています。この酵素はALOX12Bの下流で同じ脂質修飾経路に関与し、皮膚バリアの形成を助ける特殊な脂質誘導体を生成します。ALOXE3の機能喪失型変異はARCI3を引き起こし、LIとCIEの重複した臨床的特徴が見られます。ALOXE3の変異を持つ人の約3分の1は、生まれたときにコロジオン膜に包まれており、その後軽度から中等度のCIEを発症します。

ARCIの乳児は、皮膚バリアが損なわれているために多くの健康上の問題に直面します。生まれたとき、多くの赤ちゃんはコロジオン膜に包まれており、脱水症状、体温調節不全、感染症、呼吸器合併症のリスクが高まります。この膜は時間とともに剥がれ落ち、全身に広がる鱗状化や紅斑に移行します。重症型であるハーレクイン魚鱗癬では、厚い皮膚の板と深い亀裂が形成され、命に関わる合併症を引き起こす可能性があるため、集中的な新生児ケアやレチノイドなどの全身治療が必要です。
ARCIの管理は、皮膚バリアの損傷による影響を軽減することに重点を置いています。これには、高湿度環境での保湿、衛生的管理による感染予防、痛みや代謝異常のケアが含まれます。早期に保湿剤を使用することで皮膚バリアを強化し、合併症やアトピー性皮膚炎などのリスクを軽減できる可能性があります。

ARCIの軽度の型では、症状が時間とともに改善することがありますが、乾燥肌や軽度の鱗状化、角化亢進が残ることがあります。一方、ハーレクイン魚鱗癬のような重症型では、慢性的な皮膚の炎症、熱への耐性低下、関節の拘縮、眼の異常などが続くことがあります。成人では、皮膚がんのリスクが高まる可能性があり、生涯にわたる定期的な監視とケアが必要です。
ARCIの複雑性を考えると、診断のための遺伝子検査、個別化された治療計画、および根本的な遺伝子や生化学的欠陥を標的とした治療法の研究が重要です。新生児ケアの進歩と早期介入戦略により、この困難な疾患を抱える人々の予後は改善し続けています。
[10_HES7] 先天性横隔膜ヘルニア、脊肋骨異形成症(Congenital Diaphragmatic Hernia; Spondylocostal Dysostosis)

脊肋骨発育異常症タイプ4(Spondylocostal Dysostosis Type 4、SCDO4)および先天性横隔膜ヘルニア(Congenital Diaphragmatic Hernia、CDH)は、HES7遺伝子の変異に関連する稀な疾患です。この遺伝子は転写因子HES-7をコードしており、DNA転写の調節や、身体の様々な構造を形成する中胚葉という層の発育に重要な役割を果たします。HES-7は、特定のDNA領域(NボックスおよびEボックスを含むプロモーター)に作用する転写抑制因子として機能します。この機能が障害されると、重篤な発育異常を引き起こす可能性があります。
SCDO4は、胚発生中に起こる分節異常により、脊椎や肋骨の異常を特徴とする疾患です。主な特徴には、身長に比べて胴体が短いこと、首が短いこと、軽度で進行しない側弯症、脊椎の癒合、半椎骨(脊椎の一部が不完全に形成される状態)、肋骨の後方癒合、肋骨の数の減少や形態異常などがあります。新生児の場合、胸郭が小さいために呼吸機能が損なわれることがあり、重症例では集中治療を必要とすることもあります。ただし、2歳頃までに肺の成長が改善し、比較的正常な発育が可能になることもあります。それでも、重症例では慢性的な呼吸不全、肺高血圧症、心臓の問題などの合併症が起こる可能性があります。また、SCDO4の男性は鼠径ヘルニアのリスクが高く、迅速な治療が必要となる場合があります。
SCDOの診断は通常、X線撮影で行われ、脊椎の分節異常や肋骨の異常が確認されます。また、HES7を含む関連遺伝子の病的変異を特定する遺伝子検査によって確定診断が可能です。治療には、重度の側弯症に対する外科的介入、急性および慢性の呼吸不全に対する呼吸補助、成長、脊椎の湾曲、呼吸機能、神経学的問題などの定期的なモニタリングが含まれます。遺伝子カウンセリングも重要で、HES7変異によるSCDOは常染色体劣性遺伝形式で受け継がれるため、リスクのある家族に対して保因者検査や出生前診断が可能です。超音波検査では、妊娠13週頃から脊椎の異常を検出することができ、特にこの異常を詳しく調べる場合に有効です。

CDHは、HES7関連のSCDOの稀な症例で見られる横隔膜の発育異常です。横隔膜の形成や筋肉化が不完全で、横隔膜組織が欠損または不足している状態、または横隔膜が薄くなり異常に上昇する状態(挙上)として現れることがあります。この疾患はほとんどの場合、生まれた時点で明らかになり、出生1万人あたり約3〜3.6例の割合で発生します。HES7関連のSCDOでは、CDHは10%未満のケースで見られます。CDHの新生児管理では、出生直後から適切な呼吸補助が必要で、生命を脅かす呼吸不全に対応するための専門的なケアが求められます。
総じて、HES7遺伝子の変異は、初期の胚発生や転写調節が脊椎、肋骨、横隔膜の形成に密接に関与していることを示しています。これらの疾患の多様な症状や合併症に対応するためには、適切な診断、管理、カウンセリングが不可欠です。
[11_TMEM107] メッケル症候群13(Meckel syndrome 13・ジュベール症候群 Joubert Syndrome)

メッケル症候群13は、TMEM107遺伝子の変異によって引き起こされる、非常に稀で重篤な遺伝性疾患です。この疾患は常染色体劣性遺伝の形式で受け継がれます。主な特徴として、嚢胞性腎疾患、大脳奇形(特に後頭部の脳瘤)、および肝臓の線維化(管板形成異常による)があります。また、多指症(指や足の指が通常より多い状態)や小脳虫部低形成(運動調整に影響を与える可能性のある脳の異常)も見られます。残念ながら、メッケル症候群は極めて致死性が高く、多くのケースでは出生前または生後間もなく命を落とします。
TMEM107遺伝子は、トランスメンブランタンパク質107をコードしており、細胞表面に存在する「繊毛」と呼ばれる小さな毛状の構造の形成と機能において重要な役割を果たしています。この繊毛は、SHH、WNT、PDGFといった発達に関わる重要なシグナル伝達経路を調整します。これらの経路は、神経系や骨格の形成、さらには顔面や頭蓋の発達など、多くの重要なプロセスに影響を与えます。TMEM107遺伝子の変異によって繊毛の形成が障害されると、繊毛の数が減少し、異常に長くねじれた構造になるといった欠陥が生じます。この繊毛の異常が、メッケル症候群に見られる重篤な発達障害の原因となります。
メッケル症候群は、「繊毛病」と呼ばれる一群の疾患に属しており、この中にはジュベール症候群(JS)も含まれます。JSは一般的にメッケル症候群ほど重症ではなく、幼少期や成人期まで生存できる場合もありますが、脳の奇形、多指症、肝臓や腎臓の機能異常など、メッケル症候群と多くの共通点があります。これらの疾患は、TMEM107を含む繊毛関連遺伝子の変異によって引き起こされることがあります。同じ家族内で、ある個体ではメッケル症候群、別の個体ではJSが発症する例も報告されており、これらの疾患が重症度の異なるスペクトラムの一部であることが示唆されています。通常、遺伝子の機能を完全に失わせる変異はメッケル症候群を引き起こし、比較的軽微な変異はJSにつながることが多いとされています。
JSは、脳画像で見られる「モラートゥースサイン」と呼ばれる特徴的な所見、発達の遅れ、筋力低下(低緊張)、乳児期の呼吸異常などが主な特徴です。その他の症状として、視覚障害、言語の遅れ、知能の幅広い変化(正常から知的障害まで)などがあります。また、TMEM107に関連したJSの一種である「口腔顔面指症候群(JS-OFD)」では、舌の腫瘍、口蓋裂、口腔ヒダなどの顔面や口腔の異常が見られ、メッケル症候群との境界をさらに曖昧にしています。

メッケル症候群の治療は、その重篤性と高い致死率のため、主に対症療法に焦点を当てています。リスクが知られている家族では、早期診断のために非侵襲的出生前診断(NIPT)の実施が強く推奨されます。治療法は存在しないものの、関連する繊毛病(例:JS)の特定の合併症を管理するための介入が役立つ場合があります。これには、呼吸の異常、運動機能の問題、発達遅滞への治療が含まれます。また、口蓋裂、多指症、肝臓や腎臓の異常など、関連する異常に対する外科的および医療的介入が必要になることもあります。生存者においては、神経学、腎臓学、消化器学などの分野の専門家による多職種ケアが重要です。
メッケル症候群の予後は依然として厳しく、繊毛機能障害が初期発達に与える深刻な影響を反映しています。このことは、繊毛が人間の成長において果たす重要な役割と、その機能障害がもたらす重大な結果を強調しています。
【もっと知りたい方へ】Joubert Syndrome & Related Disorders Foundation
[12_CTC1] ディスケラトーシス先天性および関連するテロメア生物学的疾患(Dyskeratosis Congenita and Related Telomere Biology Disorders)

2023年に、CTC1遺伝子領域に2つの変異を持つ38歳の既婚女性がCoats plus症候群と診断された症例が報告されました。この稀な遺伝性疾患は複雑な医学的課題を伴いますが、適切な治療やケア、支援を受けることで、影響を受けた子どもたちが希望を持って生活を続けることが可能とされています。

Coats plus症候群、正式名称「脳網膜微小血管症(石灰化と嚢胞を伴う)1型(CRMCC1)」は、CTC1遺伝子の変異によるまれな劣性遺伝性疾患です。この疾患の発症には、両親からそれぞれ異常な遺伝子を1つずつ受け継ぐことが関与しています。CTC1遺伝子は、染色体の末端(テロメア)を安定させ、DNAが正確に複製されるよう助けるCST複合体という重要なタンパク質を構成しています。しかし、CTC1遺伝子に変異が生じると、この複合体が正常に機能しなくなり、テロメアが短縮し、染色体が壊れやすくなるため、病気の症状が引き起こされます。
この疾患の症状は神経系および全身に及びます。神経系では、脳内の石灰化、白質の損傷、脳嚢胞の形成が特徴的で、これにより痙性、運動失調、ジストニア、けいれん、認知機能の低下などが発生します。さらに、眼疾患として網膜血管が拡張し滲出物がたまる「Coats病」の状態がしばしば見られます。
全身的な症状としては、骨粗鬆症による骨のもろさ、消化管や肝臓の血管異常による消化管出血や門脈圧亢進症が挙げられます。また、髪や皮膚、爪に異常が見られることや、貧血や血小板減少も観察される場合があります。
この疾患は「異常角化症」など他のテロメア関連疾患と似た特徴を持つため、骨髄機能低下、がん、肺や肝臓の疾患といった合併症を早期に発見するためのスクリーニングが極めて重要です。また、Labrune症候群との鑑別には、全身症状の有無が参考になります。
CTC1遺伝子の変異に関する研究では、異常なタンパク質の生成がCST複合体の機能不全を引き起こし、テロメア短縮と染色体異常を進行させることが明らかになっています。これにより、疾患の進行や症状の悪化が生じます。
まとめると、Coats plus症候群は、眼疾患、脳の石灰化や損傷、骨のもろさ、消化管の問題など、さまざまな症状を含む複雑な疾患です。この疾患の背景には、CTC1遺伝子変異によるテロメア保護機能の障害があり、CST複合体の重要性が強調されています。
【もっと知りたい方へ】Alex, The Leukodystrophy Charity (Alex TLC)
[13_RANGRF] ブルガダ症候群(Brugada Syndrome)

ブルガダ症候群は、心臓の電気的活動に異常を引き起こす遺伝性疾患であり、生命を脅かす不整脈や突然の心停止を引き起こす可能性があります。この疾患は、主にSCN5A遺伝子の変異に関連しています。この遺伝子は、心臓の電気的リズムを維持するために重要なナトリウムチャネルであるNav1.5をコードしています。さらに、RANGRF遺伝子もこの過程において重要な役割を果たしており、MOG1というタンパク質をコードしています。このMOG1は、Nav1.5が心臓細胞の膜に正しく配置され、適切に機能するのを助ける補助因子として機能します。しかし、RANGRF遺伝子に変異が生じると、Nav1.5の膜への配置が妨げられ、ナトリウムチャネルの機能が低下します。その結果、心臓のリズムに異常が生じ、突然死のリスクが高まります。
ブルガダ症候群は通常、成人期に発症しますが、乳児期から高齢期まで報告されることがあります。突然死の平均年齢は約40歳です。この疾患の特徴的な臨床所見には、心電図(ECG)における特定のSTセグメント異常(特にV1〜V3誘導)や心室性不整脈のリスク増加が含まれます。その他の症状として、失神、乳児突然死症候群(SIDS)、および主に東南アジアで見られる突然の予期せぬ夜間死症候群(SUNDS)が挙げられます。また、男性において女性の約8倍の頻度で発症し、小児では非常に稀です。
ブルガダ症候群の診断は、特徴的な心電図所見を特定することで行われます。これは自然発生的に確認される場合もあれば、特定の薬剤を用いた誘発試験で確認される場合もあります。臨床歴や家族歴を考慮した上で、遺伝子検査によりSCN5Aまたは関連する他の遺伝子の病的変異が検出されれば、診断が確定します。
この疾患の治療は、突然死を防ぐことを目的としています。失神や心停止の既往がある患者には、植込み型除細動器(ICD)が最も効果的な治療法とされています。高リスク患者には、補助療法としてキニジンが使用される場合があり、不整脈やICDショックの発生を抑える効果があります。一部のケースでは、右心室流出路の心外膜アブレーションが有望な治療選択肢として浮上しています。また、特に家族歴や既知の遺伝子変異を持つリスクのある人々に対しては、定期的な心電図検査が推奨されます。予防策として、高熱や特定の薬剤、不整脈を誘発する可能性のある物質の回避が重要です。

ブルガダ症候群は、ほとんどの場合、常染色体優性遺伝形式で遺伝します。これは、影響を受けた親の子供が50%の確率でこの疾患を引き継ぐことを意味します。ただし、発現率の低下や症状のばらつきが見られるため、変異を持つすべての人が発症するわけではありません。稀に、ブルガダ症候群は新規(de novo)変異によって発生することもあります。影響を受けた家族に対しては、遺伝カウンセリングが推奨されており、病的変異が特定されれば、出生前または着床前の遺伝子検査も可能です。
ブルガダ症候群の世界的な有病率は約2,000人に1人と推定されていますが、地域や民族によって大きく異なります。アジアや中東地域での発症率が高く、ヒスパニック系や白人では稀とされています。多くの患者は無症状ですが、約30%が失神を経験し、8〜12%が心停止に至る可能性があります。これらのリスクを考慮すると、早期の発見と介入が非常に重要です。
[14_RPL26] ダイアモンド・ブラックファン貧血(Diamond-Blackfan Anemia)

ダイアモンド・ブラックファン貧血11(DBA11)は、RPL26遺伝子の変異によって引き起こされる稀な先天性疾患です。この遺伝子は、細胞内でタンパク質を合成するために必要なリボソームの一部を作る役割を持っています。RPL26の機能が失われると、リボソームの働きが障害され、結果としてDBA11が発症します。DBA11はダイアモンド・ブラックファン貧血(DBA)の一つのタイプで、生後1年以内に現れる再生不良性の貧血を特徴としています。
DBAの主な特徴は中等度から重度の貧血で、しばしば赤血球が通常より大きくなる(大球性)状態がみられます。また、骨髄内で赤血球を作り出す細胞(赤血球前駆細胞)が大幅に減少する一方、白血球や血小板の数は通常正常です。患者の30〜40%が成長障害や先天性異常を伴い、顔の形状異常(ピエール・ロバン症候群や口蓋裂)、親指の異常、泌尿生殖器の異常などが含まれます。重症例では、胎児の貧血によって胎児水腫が生じることもあります。また、DBAは特定のがん、例えば急性骨髄性白血病(AML)、骨髄異形成症候群(MDS)、骨肉腫などのリスクを高めることが知られています。
診断は、生後1年以内に発症する大球性貧血、未熟赤血球(網赤血球)の減少、骨髄内の赤血球前駆細胞の不足、そして他の原因が見つからない場合に行われます。遺伝学的診断では、DBAに関連する病的な遺伝子変異を特定することで確定されます。多くの場合、DBAは親から引き継がれる常染色体優性遺伝の形をとりますが、一部ではGATA1やTSR2という遺伝子の変異によるX連鎖性遺伝も報告されています。
治療の中心は、貧血の改善と合併症の予防です。12か月以上の子どもには副腎皮質ステロイド剤が効果的な場合が多いですが、長期使用は感染症、成長抑制、骨密度の低下といった副作用を引き起こす可能性があります。ステロイドが効かない場合や、副作用が強い場合には、赤血球輸血や造血幹細胞移植(HSCT)が必要になることがあります。HSCTは、DBAによる貧血を根本的に治療する唯一の方法です。また、骨や目、内分泌に関連する問題については、専門医と連携して治療が行われます。がんの治療では、血液細胞の減少を防ぐため、化学療法の使用には慎重な判断が必要です。

輸血による鉄過剰症などの二次的な合併症には、鉄キレート療法が用いられます。輸血による鉄過剰やステロイドの副作用を定期的にチェックすることが重要です。経過観察では、血液検査や骨髄評価、がんスクリーニングが行われます。また、分子遺伝学的検査によってリスクのある家族を特定し、早期対応を図ることも可能です。
DBAは主に常染色体優性遺伝により受け継がれ、患者の40〜45%が親から病的変異を引き継いでいます。残りのケースは新規の変異によるものです。X連鎖性の場合、変異を持つ男性はその変異をすべての娘に伝えますが、息子には伝わりません。女性が変異を持つ場合、子どもに変異が伝わる確率は50%で、息子が変異を受け継ぐと発症し、娘の場合は通常キャリアとなります。
妊娠中には母体のヘモグロビン値を注意深くモニタリングし、必要に応じて低用量のアスピリンを使用することで合併症リスクを軽減することができます。遺伝カウンセリングは、影響を受けた家族に推奨されており、出生前診断や着床前診断も選択肢の一つです。
DBAは、細胞のリボソーム機能と全身の発達過程がいかに密接に関連しているかを示しています。リボソームの障害によって、血液の異常や全身の先天性異常が引き起こされることが理解されています。
[15_NTN1] 先天性鏡動作(Congenital Mirror Movements)

ミラームーブメント4(Mirror Movements 4, MRMV4)は、NTN1遺伝子の変異によって引き起こされるまれな遺伝性疾患です。この遺伝子は、神経軸索の誘導と細胞の生存に重要なタンパク質「ネトリン-1」をコードしています。ネトリン-1は中枢神経系の交連軸索(左右の神経を結ぶ軸索)や末梢運動軸索の成長を導きます。ネトリン-1は、DCCやUNC5ファミリーと呼ばれる特定の受容体と結合することで、軸索の「引き寄せ」や「排斥」を調整します。たとえば、UNC5Cという受容体に結合すると、この受容体が細胞骨格の構成要素である微小管から分離され、微小管の動きが活発化し、軸索の排斥が促されます。また、ネトリン-1はアポトーシス(プログラム細胞死)を防ぐことで神経細胞の生存を助ける役割もあり、これが腫瘍の発生や進行に関連していることもあります。
ネトリン-1の機能が失われると、MRMV4が発症します。この疾患は常染色体優性遺伝の形式で遺伝し、体の片側の随意運動が反対側で鏡のように反射される「ミラームーブメント」と呼ばれる非自発的な動きを特徴とします。幼児期には一時的なミラームーブメントが見られることがありますが、7歳を過ぎても消えない場合は異常であり、病的と考えられます。MRMV4では、これらの動きは主に上肢に現れ、特に指や手の動きが最も強く影響を受けます。通常、歩行には支障がありませんが、足の指の筋肉が軽度に影響を受けることがあります。
先天性のミラームーブメントは通常、幼少期に現れ、成人期を通じて進行や改善のないまま持続します。これらの動きの強さには個人差がありますが、意図的な動きよりも通常は弱くなります。一部の人では、繰り返しの手作業や持続的な動作によって上肢に痛みを感じることがあり、細かい作業や片手だけでの動作を必要とする日常生活のタスクに支障をきたすことがあります。脳のMRI検査では通常、異常が見られませんが、DCC遺伝子の変異による場合、脳梁の部分的または完全な欠如が認められることがあります。一方、これまでのところNTN1遺伝子の変異によるケースで脳梁異常が報告されたことはありません。

この疾患の治療は、進行性ではないため適応に重点を置きます。たとえば、子どもには試験時間を延長したり、筆記作業を減らしたりといった教育面での配慮が推奨されます。青年期や成人期には、複雑な両手の協調動作や長時間の手作業を必要としない職業を選ぶことが勧められます。また、社会的な偏見を防ぎ、本人の能力を最大限に引き出すために心理的なサポートも重要です。
先天性のミラームーブメントは、常染色体優性遺伝の形式で受け継がれますが、発症の確率(浸透率)が低い場合があります。このため、症状がある親から遺伝子変異を受け継いだとしても、必ずしも発症するとは限りません。また、発症していない親から兄弟姉妹に遺伝する可能性もあります。原因となる遺伝子変異が特定された場合、出生前診断や着床前診断を選択肢とすることが可能です。
この疾患の有病率は非常に低く、推定1,000,000人に1人未満とされています。ただし、軽症例の未診断が多いため、実際の有病率はこれより高い可能性があります。ミラームーブメントの主な原因は、NTN1、DCC、またはRAD51の変異ですが、最も一般的なのはDCCに関連するケースで、発症の確率は約42%とされています。
将来の見通し
DNAシーケンシング技術の進歩により、非侵襲的出生前診断(NIPT)は、出生前FISH(蛍光in situハイブリダイゼーション)や羊水検査のような侵襲的な方法に代わる信頼性が高く安全なスクリーニング方法として登場しました。羊水検査では、子宮に針を刺して羊水や絨毛膜の細胞を採取しますが、NIPTは母親の血液に含まれる胎児の細胞を分析することで、母体や胎児の健康にリスクを与えることなく、信頼できる結果を提供します。このため、NIPTは安全なスクリーニング方法を求める妊婦さんたちの間で、ますます人気のある選択肢となっています。
たとえ遺伝的な異常が見つかったとしても、多くの場合、その子どもが成長し、実りある人生を送るための道や希望は残されています。特に研究が進展している今、その可能性はますます広がっています。NIPTによる早期発見が、不安の原因ではなく、安心を得る手段となり、ご家族の未来に備えるための一助となることを願っています。

引用文献
- Méneret A, Trouillard O, Dunoyer M, et al. Congenital Mirror Movements. 2015 Mar 12 [Updated 2020 Sep 24]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279760/
- Janov, A. J., Leong, T., Nathan, D. G., & Guinan, E. C. (1996). Diamond-Blackfan anemia. Natural history and sequelae of treatment. Medicine, 75(2), 77–78. https://doi.org/10.1097/00005792-199603000-00004
- Willig, T.-N., Niemeyer, C. M., Leblanc, T., Tiemann, C., Robert, A., Budde, J., Lambiliotte, A., Kohne, E., Souillet, G., Eber, S., Stephan, J.-L., Girot, R., Bordigoni, P., Cornu, G., Blanche, S., Guillard, J. M., & Mohandas, N. (1999). Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 diamond-blackfan anemia patients. Pediatric Research, 46(5), 553–553. https://doi.org/10.1203/00006450-199911000-00011
- Sieff C. Diamond-Blackfan Anemia. 2009 Jun 25 [Updated 2023 Mar 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK7047/
- Marfatia, K. A., Harreman, M. T., Fanara, P., Vertino, P. M., & Corbett, A. H. (2001). Identification and characterization of the human MOG1 gene. Gene, 266(1-2), 45–56. https://doi.org/10.1016/s0378-1119(01)00364-x
- Brugada R, Campuzano O, Sarquella-Brugada G, et al. Brugada Syndrome. 2005 Mar 31 [Updated 2022 Aug 25]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1517/
- Campuzano, O., Berne, P., Selga, E., Allegue, C., Iglesias, A., Brugada, J., & Brugada, R. (2014). Brugada syndrome and p.E61X_RANGRF. Cardiology journal, 21(2), 121–127. https://doi.org/10.5603/CJ.a2013.0125
- Kayarian, F. B., Cohen, S. M., Cohen, M. L., & Sammartino, D. E. (2023). Coats plus syndrome presenting in an adult. Journal of VitreoRetinal Diseases, 7(6), 562–564. https://doi.org/10.1177/24741264231171465
- Anderson, B. H., Kasher, P. R., Mayer, J., Szynkiewicz, M., Jenkinson, E. M., Bhaskar, S. S., Urquhart, J. E., Daly, S. B., Dickerson, J. E., O’Sullivan, J., Leibundgut, E. O., Muter, J., Abdel-Salem, G. M., Babul-Hirji, R., Baxter, P., Berger, A., Bonafé, L., Brunstom-Hernandez, J. E., Buckard, J. A., Chitayat, D., … Crow, Y. J. (2012). Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nature genetics, 44(3), 338–342. https://doi.org/10.1038/ng.1084
- Polvi, A., Linnankivi, T., Kivelä, T., Herva, R., Keating, J. P., Mäkitie, O., Pareyson, D., Vainionpää, L., Lahtinen, J., Hovatta, I., Pihko, H., & Lehesjoki, A. E. (2012). Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. American journal of human genetics, 90(3), 540–549. https://doi.org/10.1016/j.ajhg.2012.02.002
- Cela, P., Hampl, M., Shylo, N. A., Christopher, K. J., Kavkova, M., Landova, M., Zikmund, T., Weatherbee, S. D., Kaiser, J., & Buchtova, M. (2018). Ciliopathy Protein Tmem107 Plays Multiple Roles in Craniofacial Development. Journal of dental research, 97(1), 108–117. https://doi.org/10.1177/0022034517732538
- Lambacher, N. J., Bruel, A. L., van Dam, T. J., Szymańska, K., Slaats, G. G., Kuhns, S., McManus, G. J., Kennedy, J. E., Gaff, K., Wu, K. M., van der Lee, R., Burglen, L., Doummar, D., Rivière, J. B., Faivre, L., Attié-Bitach, T., Saunier, S., Curd, A., Peckham, M., Giles, R. H., … Blacque, O. E. (2016). TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nature cell biology, 18(1), 122–131. https://doi.org/10.1038/ncb3273
- Parisi M, Glass I. Joubert Syndrome. 2003 Jul 9 [Updated 2017 Jun 29]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1325/
- Turnpenny PD, Sloman M, Dunwoodie S. Spondylocostal Dysostosis, Autosomal Recessive. 2009 Aug 25 [Updated 2023 Aug 17]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK8828/
- Longoni M, Pober BR, High FA. Congenital Diaphragmatic Hernia Overview. 2006 Feb 1 [Updated 2020 Nov 5]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1359/
- Oji, V., Preil, M. L., Kleinow, B., Wehr, G., Fischer, J., Hennies, H. C., Hausser, I., Breitkreutz, D., Aufenvenne, K., Stieler, K., Tantcheva-Poór, I., Weidinger, S., Emmert, S., Hamm, H., Perusquia-Ortiz, A. M., Zaraeva, I., Diem, A., Giehl, K., Fölster-Holst, R., Kiekbusch, K., … Traupe, H. (2017). S1 guidelines for the diagnosis and treatment of ichthyoses – update. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG, 15(10), 1053–1065. https://doi.org/10.1111/ddg.13340
- Egawa, G., & Kabashima, K. (2018). Barrier dysfunction in the skin allergy. Allergology International, 67(1), 3–11. https://doi.org/10.1016/j.alit.2017.10.002
- Richard G. Autosomal Recessive Congenital Ichthyosis. 2001 Jan 10 [Updated 2023 Apr 20]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1420/
- Yang, P., Pardon, L. P., Ho, A. C., Lauer, A. K., Yoon, D., Boye, S. E., Boye, S. L., Roman, A. J., Wu, V., Garafalo, A. V., Sumaroka, A., Swider, M., Viarbitskaya, I., Aleman, T. S., Pennesi, M. E., Kay, C. N., Fujita, K. P., & Cideciyan, A. V. (2024). Safety and efficacy of ATSN-101 in patients with Leber congenital amaurosis caused by biallelic mutations in GUCY2D: A phase 1/2, multicentre, open-label, unilateral dose escalation study. The Lancet, 404(10456), 962–970. https://doi.org/10.1016/S0140-6736(24)01447-8
- Tsang, S. H., & Sharma, T. (2018). Leber congenital amaurosis. In S. H. Tsang & T. Sharma (Eds.), Atlas of Inherited Retinal Diseases (Vol. 1085, pp. 131–137). Springer International Publishing. https://doi.org/10.1007/978-3-319-95046-4_26
- Kumaran N, Pennesi ME, Yang P, et al. Leber Congenital Amaurosis / Early-Onset Severe Retinal Dystrophy Overview. 2018 Oct 4 [Updated 2023 Mar 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK531510/
- Lin, M.-H., Chou, P.-C., Lee, I.-C., Yang, S.-F., Yu, H.-S., & Yu, S. (2023). Inherited reticulate pigmentary disorders. Genes, 14(6), 1300. https://doi.org/10.3390/genes14061300
- Savage SA, Niewisch MR. Dyskeratosis Congenita and Related Telomere Biology Disorders. 2009 Nov 12 [Updated 2023 Jan 19]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK22301/
- Turner JT, Brzezinski J, Dome JS. Wilms Tumor Predisposition. 2003 Dec 19 [Updated 2022 Mar 24]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1294/
- Schneider K, Zelley K, Nichols KE, et al. Li-Fraumeni Syndrome. 1999 Jan 19 [Updated 2024 Sep 5]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1311/
- Van Tol, W., Ashikov, A., Korsch, E., Abu Bakar, N., Willemsen, M. A., Thiel, C., & Lefeber, D. J. (2019). A mutation in mannose‐phosphate‐dolichol utilization defect 1 reveals clinical symptoms of congenital disorders of glycosylation type I and dystroglycanopathy. JIMD Reports, 50(1), 31–39. https://doi.org/10.1002/jmd2.12060
- Sparks SE, Krasnewich DM. Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview. 2005 Aug 15 [Updated 2017 Jan 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1332/
- Thompson, R., Bonne, G., Missier, P., & Lochmüller, H. (2019). Targeted therapies for congenital myasthenic syndromes: systematic review and steps towards a treatabolome. Emerging topics in life sciences, 3(1), 19–37. https://doi.org/10.1042/ETLS20180100
- Abicht A, Müller JS, Lochmüller H. Congenital Myasthenic Syndromes Overview. 2003 May 9 [Updated 2021 Dec 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1168/
- Degtyareva, A. V., Nikitina, I. V., Orlovskaya, I. V., Zakharova, E. Y., Baidakova, G. V., Ionov, O. V., … & Levadnaya, A. V. (2016). Very long-chain acyl-coenzyme A dehydrogenase deficiency. Rossiyskiy Vestnik Perinatologii i Pediatrii (Russian Bulletin of Perinatology and Pediatrics), 59(4), 41-47.
- Mendez-Figueroa, H., Shchelochkov, O. A., Shaibani, A., Aagaard-Tillery, K., & Shinawi, M. S. (2010). Clinical and biochemical improvement of very long-chain acyl-CoA dehydrogenase deficiency in pregnancy. Journal of perinatology : official journal of the California Perinatal Association, 30(8), 558–562. https://doi.org/10.1038/jp.2009.198
- Leslie ND, Saenz-Ayala S. Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. 2009 May 28 [Updated 2023 Jul 13]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK6816/
- Rodríguez-Palmero, A., Boerrigter, M. M., Gómez-Andrés, D., Aldinger, K. A., Marcos-Alcalde, Í., Popp, B., Everman, D. B., Lovgren, A. K., Arpin, S., Bahrambeigi, V., Beunders, G., Bisgaard, A. M., Bjerregaard, V. A., Bruel, A. L., Challman, T. D., Cogné, B., Coubes, C., de Man, S. A., Denommé-Pichon, A. S., Dye, T. J., … Tümer, Z. (2021). DLG4-related synaptopathy: a new rare brain disorder. Genetics in medicine : official journal of the American College of Medical Genetics, 23(5), 888–899. https://doi.org/10.1038/s41436-020-01075-9
- Tümer Z, Dye TJ, Prada C, et al. DLG4-Related Synaptopathy. 2023 Jun 22. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK592682/
- Perez, G., Barber, G. P., Benet-Pages, A., Casper, J., Clawson, H., Diekhans, M., Fischer, C., Gonzalez, J. N., Hinrichs, A. S., Lee, C. M., Nassar, L. R., Raney, B. J., Speir, M. L., van Baren, M. J., Vaske, C. J., Haussler, D., Kent, W. J., & Haeussler, M. (2024). The UCSC Genome Browser database: 2025 update. Nucleic Acids Research, gkae974. https://doi.org/10.1093/nar/gkae974
- Harrison, P. W., Amode, M. R., Austine-Orimoloye, O., Azov, A. G., Barba, M., Barnes, I., Becker, A., Bennett, R., Berry, A., Bhai, J., Bhurji, S. K., Boddu, S., Branco Lins, P. R., Brooks, L., Budhanuru Ramaraju, S., Campbell, L. I., Carbajo Martinez, M., Charkhchi, M., Chougule, K., … Yates, A. D. (2024). Ensembl 2024. Nucleic Acids Research, 52(D1), D891–D899. https://doi.org/10.1093/nar/gkad1049
- Jumper, J et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021).
- Varadi, M et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research (2021).
- Cheng, J et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science (2023).







