




スミス・マジェニス症候群(SMS)の詳細な説明。症状、診断方法、治療法、そして患者の将来の見通しについて知り、早期介入の重要性や遺伝子検査について理解を深めましょう。

ダウン症の検査
気になる費用はこちら
この記事のまとめ
スミス・マジェニス症候群(SMS)は、認知障害、行動的課題、そして顕著な睡眠障害を特徴とする複雑な遺伝性疾患です。この記事では、SMSの症状、診断方法、治療法について説明し、早期診断と適切な支援が患者の生活の質を向上させる可能性を探ります。
疾患概要


スミス・マジェニス症候群(SMS)は、複数の身体システムに影響を与える稀な遺伝性の神経発達障害です。この症候群は、さまざまな程度の認知障害、行動の異常、そして顕著な睡眠障害を特徴としています。SMSの患者は、年齢とともに特徴的な身体的特徴がさらに明確になり、心臓や腎臓などに関連するさまざまな先天的異常も見られます。
乳児期のSMSの一般的な兆候としては、授乳の困難、成長不良、筋力低下(低筋緊張)、反射の遅れが含まれます。また、過度の眠気を感じることがあり、授乳のために頻繁に目を覚ます必要があったり、全体的に元気がないといった症状も見られます。成長するにつれて、SMSの子どもたちは発達の遅れ、知的障害、さまざまな行動の問題を抱えることが一般的です。これには、顕著な睡眠障害が含まれます。睡眠の問題は、メラトニンの逆転したサーカディアンリズム(生体リズム)が関係しており、睡眠パターンが乱れることがあります。その他の一般的な行動問題には、ステレオタイプ行動や不適応行動、自己傷害(自分を叩く、自分を噛むなど)、強い不安感が含まれます。注意力の欠如、気が散りやすさ、過活動、衝動的な行動といった実行機能の問題もよく見られます。また、多くの人々は特定の質感や音、経験に対して過敏に反応したり、逆にそれらを繰り返し求める傾向もあります。トイレに関する問題も一般的です。

SMSの特徴的な身体的特徴は、年齢を重ねるにつれてより顕著になります。これには、年齢とともに進行する粗い顔の特徴が含まれます。他の身体的異常には、歯の問題、小さな骨の異常、目、耳、喉の異常が見られることがあります。声がかすれていることや、早期の表現言語の遅れ(聴力に問題がある場合もある)もよく見られます。さらに、SMSの患者はしばしば短い身長や、乳児期に成長不良を示すことがあります。
スミス・マジェニス症候群の発症率は、出生1万人あたり25人程度とされていますが、最近の診断技術の進歩により、実際の有病率は1万人あたり15人に近い可能性があるとされています。正確な有病率は完全には分かっていませんが、過去に報告されていなかったケースもあるため、実際の発症率は現在の推定値よりも高い可能性があります。
SMSは、主に17番染色体のp11.2領域にあるインタースティシャル欠失によって引き起こされ、これによりRAI1遺伝子が不十分に発現します。この遺伝子は、症候群の特徴に重要な役割を果たしています。欠失がない場合でも、RAI1遺伝子の変異によって、同じ症候群の特徴が現れることがあります。診断は、身体的、発達的、行動的な特徴の独特なパターンを認識することで行われます。これらの特徴の多くは幼少期には目立たず、年齢とともにより明確になります。
知的機能の程度は個人によって異なり、多くの人々は軽度から中度の知的障害を持っています。SMSは多系統にわたる疾患であり、特に神経行動学的な特徴が顕著です。患者の感情的および社会的発達は、知的成熟に比べて遅れることが多いです。治療と管理には、身体的、発達的、行動的なさまざまな課題に対応するための多面的なアプローチが必要です。
病因と診断の方法
スミス・マジェニス症候群(SMS)は、17番染色体の17p11.2領域に欠失があるか、またはその領域にあるレチノイン酸誘導遺伝子1(RAI1)に病的な変異があることが原因で起こる複雑な遺伝性疾患です。SMSの約90%はこの染色体領域の欠失が原因で、最も一般的なのはRAI1遺伝子に影響を与える3.7Mbの欠失です。この欠失により、RAI1タンパク質が十分に作られなくなり、神経発達や行動機能、体内時計(昼夜のリズム)などに必要な働きが不足します。約10%の症例では、17p11.2領域の欠失は見られませんが、RAI1遺伝子に直接的な病的変異が見つかります。
RAI1遺伝子は、細胞内での遺伝子の働きを調整する重要な役割を担っており、その不足がSMSの主な特徴である発達遅延や知的障害、特有の行動的特徴を引き起こします。行動面では、特に顕著な睡眠障害、決まった行動パターン(ステレオタイプ行動)、自己傷害、不適応行動などが見られます。これらは子どもが成長するにつれてより目立つようになり、多くの症状は18ヶ月頃に明確になることが多いです。
SMSの診断は、身体的、発達的、行動的な特徴をもとに行われます。これらの特徴は幼少期には微妙に現れ、年齢を重ねるごとに顕著になります。SMSの患者は、粗い顔つきなどの特徴的な顔貌を持ち、年齢とともにその特徴がより明確になります。加えて、歯の問題、軽い骨の異常、目や耳、喉に関する問題(例えばかすれた声)が見られることがあります。言語の発達遅延や表現力の遅れも一般的で、聴力に問題がある場合もあります。
身長や体重は、遺伝的な異常によって異なることがあります。17p11.2領域の欠失がある患者は、RAI1変異の患者よりも短身の割合が高く、17p11.2の欠失がある81%の患者は短身であるのに対し、RAI1変異がある患者ではその割合は10%程度です。
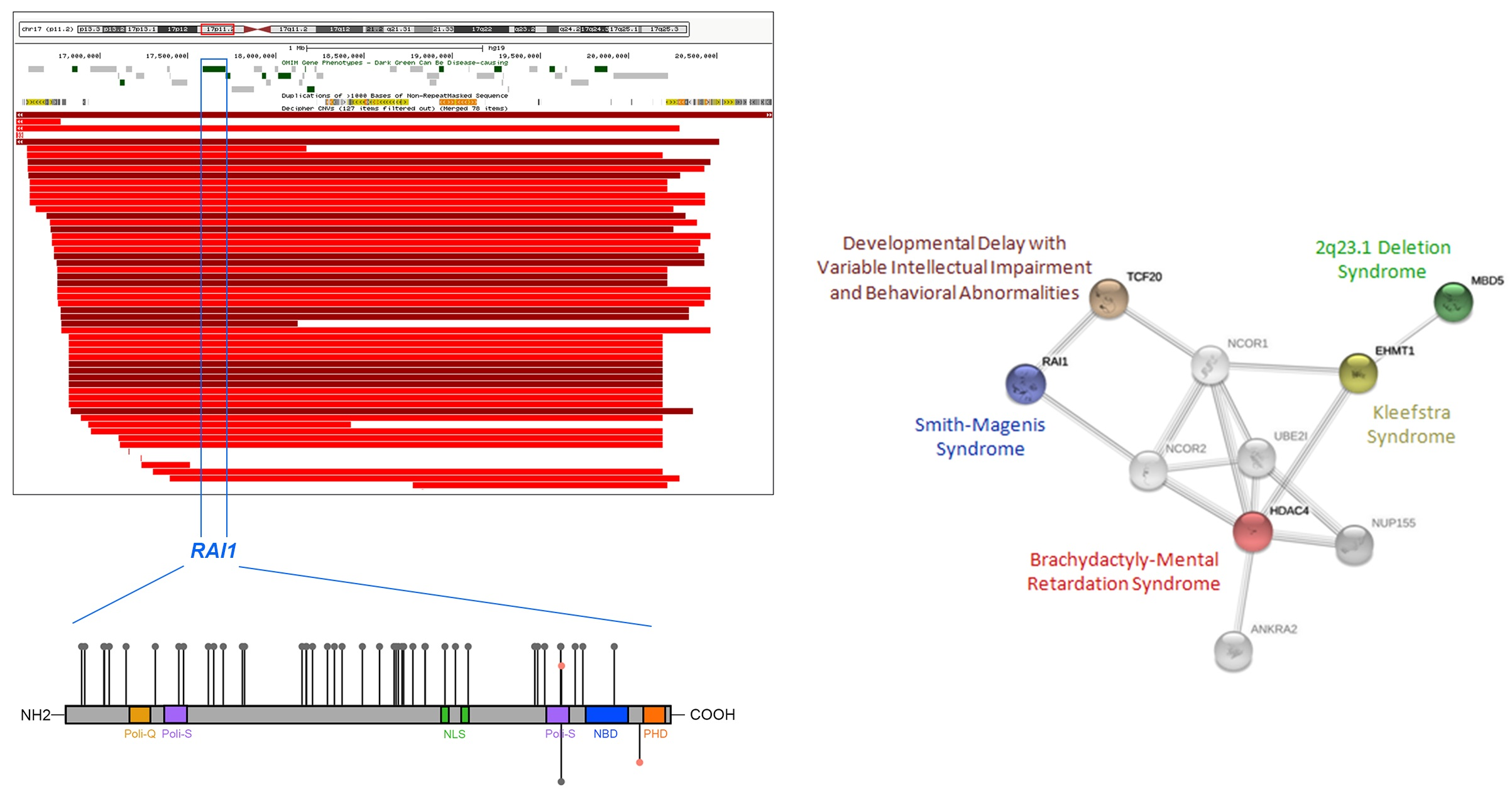
Rinaldi, B., Villa, R., Sironi, A., Garavelli, L., Finelli, P., & Bedeschi, M. F. (2022). Smith-Magenis Syndrome—Clinical Review, Biological Background and Related Disorders. Genes, 13(2), 335. https://doi.org/10.3390/genes13020335
SMSは通常、親から遺伝することなく新たに発生した遺伝子変異が原因となりますが、まれに家族内で遺伝することもあります。SMSの最も一般的な遺伝的原因は17p11.2領域の欠失で、1.5Mbから9Mbの大きさがあります。この欠失は、非対立的相同組換え(NAHR)と呼ばれるメカニズムによって引き起こされ、繰り返し配列が多く含まれているこの領域では欠失の大きさにばらつきが見られます。
17p11.2領域には約80個の遺伝子が存在しますが、RAI1がSMSの原因遺伝子として特定されて以来、典型的な17p11.2の欠失が見られない症例でもRAI1の病的変異が原因でSMSが発症することが明らかになり、これまでより多くの症例が発見されています。
SMSは常染色体優性遺伝と考えられ、通常は新たに発生した遺伝子変異が原因ですが、まれに影響を受けた親から遺伝することもあります。
疾患の症状と管理方法
スミス・マジェニス症候群(SMS)は、身体的、発達的、行動的な面でさまざまな症状を引き起こす遺伝性疾患です。SMSに対する特効薬はありませんが、治療は、個々の患者が抱えるさまざまな医療的および発達的な課題に対処することを中心に行われます。早期介入や個別化された治療戦略が、健康や生活の質、社会的機能の向上に重要です。
SMSのほとんどの患者は行動の問題や睡眠障害を抱えているため、治療にはこれらの基本的な症状に対処するための行動療法や教育的介入がよく行われます。精神薬を使用する場合もありますが、SMSにおけるその効果に関する証拠は限られています。治療に使用される薬は、臨床経験やケーススタディをもとに選ばれることが多く、一般的には自閉症やADHDに関連する症状を管理することを目的としています。

過活動やそれに関連する行動には、メチルフェニデート(MPH)などの刺激薬がよく使用されます。これらは効果的で耐容性も高いため、第一選択薬とされています。また、クロンジンという薬も、刺激薬と併用または代替として使用されることがあり、これにより睡眠が改善されることもあります。抗精神病薬の一つであるリスペリドンは、過活動、攻撃性、自己傷害などの行動に対処するためによく処方されますが、体重増加や食欲増進、疲労などの副作用が見られることがあります。
睡眠障害もSMSの重要な課題です。規則正しい生活リズムを保ち、リラックスできる寝室を作り、カフェインなどの刺激物を避けるといった睡眠衛生の改善が推奨されますが、睡眠障害の管理には薬物療法がしばしば必要です。メラトニンは、SMSに見られる体内時計の乱れを改善するためによく使用されます。短期間のメラトニン補充により、入眠までの時間が短縮されることが示されていますが、全体的な睡眠時間や行動への影響はあまり見られません。一部の患者では、メラトニンとアセブトロールなどの薬を組み合わせることで、睡眠と行動の改善が見られることがあります。
タシメルテオンというメラトニン受容体アゴニストは、SMSにおける睡眠障害の治療のためにアメリカで承認されています。その他、抗ヒスタミン薬や抗うつ薬、抗けいれん薬も、睡眠問題やけいれんを伴う症状の治療に役立つ場合があります。

SMSは、中程度から重度の知的障害、言語遅延、さまざまな行動的な課題を特徴としています。攻撃的な行動はSMSの患者に一般的で、38%から93%の患者が見られます。自己傷害行動(自分を叩く、かむ、皮膚を引っ掻くなど)は、70%から97%の患者に見られます。これらの行動は他の発達障害で見られるものに似ていることもありますが、目を突いたり、壁を打ち破ったり、爪を引っこ抜いたりといった珍しい行動もあります。これらの行動は成人の注意を引くために起こることが多いと考えられていますが、その原因についてはまだ調査が進んでいません。
SMSの子どもが成長するにつれて、行動の特徴はより顕著になり、社会的な交流の難しさ、衝動性、非適応的行動を通じて注意を引こうとする傾向が見られます。睡眠障害は、これらの行動の予測因子として強い関連があります。
SMSには、発育の遅れを含む身体的な問題も多くあります。短身や乳児期の授乳困難などの成長障害が一般的です。肥満は、通常、子どもの頃から始まり、成人期には健康リスクを高める原因となることがあります。胃食道逆流症や便秘などの消化器系の問題も頻繁に報告されています。また、歯の異常や口の機能に関する問題、側弯症や異常な歩行など、運動器系の問題も見られます。

感覚の統合に問題を抱えることもあり、音に敏感であったり、感覚情報をうまく統合できなかったりすることがあります。聴力の問題、特に伝音性難聴や聴力の変動がよく見られ、頻繁に耳の感染症の治療が必要になります。目の問題、例えば斜視や近視もよく見られ、年齢とともに網膜剥離を経験することがあるかもしれません。
心血管系や腎臓に関する問題がSMS患者に発生することもありますが、必ずしも全員に現れるわけではありません。心臓の欠陥(弁の問題や中隔欠損症など)や腎臓の奇形が見られることがあります。内分泌系の問題(甲状腺機能低下症など)も一般的で、思春期の遅れや早期の性成熟も観察されています。

SMSの治療には、医療面だけでなく、発達支援も重要です。早期の介入プログラム、特別支援教育、言語療法などが効果的です。家族への心理社会的サポートも重要で、介護の負担を軽減するための支援が求められます。
SMSには継続的な監視と支援が必要です。発育、睡眠、行動の課題を追跡し、必要な医療や教育支援を提供するために定期的な評価が推奨されます。定期的な聴力、視力、健康チェックも必要です。新たな合併症が発生していないかを確認し、成長や栄養状態も監視します。
適切な治療と個別対応により、多くのSMS患者は充実した生活を送ることができます。
将来の見通し
スミス・マジェニス症候群(SMS)は複雑な遺伝性疾患であり、生命予後に関するデータは限られていますが、重大な臓器障害がない場合、SMSのある人々は一般的に知的障害がある人々と同じような寿命を持つと考えられています。最も高齢のSMS患者は88歳まで生きたことが知られており、彼女の最後の数ヶ月は、睡眠の問題や慢性的な副鼻腔炎に悩まされながらも、依然として元気で幸せそうだったと報告されています。死の数日前に脳卒中を発症しましたが、詳細を調べるための解剖は行われませんでした。この事例は、適切なケアがあればSMSのある人々も長生きできる可能性があることを示唆していますが、長期的な影響には不確定な点もあります。

SMSの症状の重症度や範囲は個人によって大きく異なるため、多職種によるアプローチが不可欠です。内分泌学、遺伝学、神経学などの専門家が協力して、個々のニーズに対応することが重要です。早期診断は、適切な管理と介入を確実に行うために非常に重要です。遺伝子検査、特に染色体マルチアレイ検査は、臨床治療と研究の両方において有用です。DNAシーケンシング技術の進歩により、非侵襲的な出生前診断(NIPT)がSMSを出生前に検出するための信頼性が高く、安全な選択肢となっています。羊水検査や絨毛検査などの侵襲的な方法とは異なり、NIPTは母親の血液中に含まれる胎児細胞を分析することで、母体や胎児の健康にリスクを与えることなく正確な結果を提供します。このため、妊娠中の安全なスクリーニング方法を求める親にとって魅力的な選択肢となっています。
SMSがもたらす課題にもかかわらず、早期かつ正確な診断により、個々の生活の質や全体的な予後が大きく改善される可能性があります。タイムリーな介入とサポートを受けることで、SMSを持って生まれた子どもたちも充実した生活を送ることができます。SMSを持つすべての子どもたちには希望があり、適切なケアを受けることで、その潜在能力を最大限に引き出すことができます。
もっと知りたい方へ
【支援団・写真あり・英語】PRISMS | Parents and Researchers Interested in Smith-Magenis Syndrome
PRISMS(Smith-Magenis Syndrome Advocacy、Education、Support Organization)は、スミス・マジェニス症候群(SMS)を持つ個人、その家族、そして彼らを支援する専門家のための支援団体です。PRISMSは、教育、認知向上、そしてSMSに影響を受けたすべての人々のための研究機会を創出することを目的として、1993年2月4日に親と専門家のグループによって設立された、非営利の501(c)(3)団体です。
引用文献
- Smith ACM, Boyd KE, Brennan C, et al. Smith-Magenis Syndrome. 2001 Oct 22 [Updated 2022 Mar 10]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1310/
- Hicks, M., Ferguson, S., Bernier, F., & Lemay, J. F. (2008). A case report of monozygotic twins with Smith-Magenis syndrome. Journal of developmental and behavioral pediatrics : JDBP, 29(1), 42–46. https://doi.org/10.1097/DBP.0b013e318146903d
- Rinaldi, B., Villa, R., Sironi, A., Garavelli, L., Finelli, P., & Bedeschi, M. F. (2022). Smith-magenis syndrome—Clinical review, biological background and related disorders. Genes, 13(2), 335. https://doi.org/10.3390/genes13020335
- Boot, E., Linders, C. C., Tromp, S. H., van den Boogaard, M. J., & van Eeghen, A. M. (2021). Possible underreporting of pathogenic variants in RAI1 causing Smith-Magenis syndrome. American journal of medical genetics. Part A, 185(10), 3167–3169. https://doi.org/10.1002/ajmg.a.62380
- Sloneem, J., Oliver, C., Udwin, O., & Woodcock, K. A. (2011). Prevalence, phenomenology, aetiology and predictors of challenging behaviour in Smith‐Magenis syndrome. Journal of Intellectual Disability Research, 55(2), 138–151. https://doi.org/10.1111/j.1365-2788.2010.01371.x
- Smith, A. C. M., & Gropman, A. L. (2021). Smith–magenis syndrome. In J. C. Carey, A. Battaglia, D. Viskochil, & S. B. Cassidy (Eds.), Cassidy and Allanson’s Management of Genetic Syndromes (1st ed., pp. 863–893). Wiley. https://doi.org/10.1002/9781119432692.ch54
- Orphanet. (Last updated November 2020). Smith-Magenis syndrome. Reviewed by Dr Laurence PERRIN. Retrieved from https://www.orpha.net/en/disease/detail/819
- Online Mendelian Inheritance in Man. SMITH-MAGENIS SYNDROME; SMS (Last updated May 2017 by Marla J. F. O’Neill). Retreived from https://omim.org/entry/182290
- Perez, G., Barber, G. P., Benet-Pages, A., Casper, J., Clawson, H., Diekhans, M., Fischer, C., Gonzalez, J. N., Hinrichs, A. S., Lee, C. M., Nassar, L. R., Raney, B. J., Speir, M. L., van Baren, M. J., Vaske, C. J., Haussler, D., Kent, W. J., & Haeussler, M. (2024). The UCSC Genome Browser database: 2025 update. Nucleic Acids Research, gkae974. https://doi.org/10.1093/nar/gkae974
- Harrison, P. W., Amode, M. R., Austine-Orimoloye, O., Azov, A. G., Barba, M., Barnes, I., Becker, A., Bennett, R., Berry, A., Bhai, J., Bhurji, S. K., Boddu, S., Branco Lins, P. R., Brooks, L., Budhanuru Ramaraju, S., Campbell, L. I., Carbajo Martinez, M., Charkhchi, M., Chougule, K., … Yates, A. D. (2024). Ensembl 2024. Nucleic Acids Research, 52(D1), D891–D899. https://doi.org/10.1093/nar/gkad1049
スミス・マジェニス症候群(SMS)の詳細な説明。症状、診断方法、治療法、そして患者の将来の見通しについて知り、早期介入の重要性や遺伝子検査について理解を深めましょう。
NIPT(新型出生前診断)について詳しく見る
関連記事
-
NEW

-

-

-

-

-








